v1.1 Cheat Sheet
Sequence → physicochemical scales → interpretable features → explainable ML → biological mechanism
pip install aaanalysis · aaanalysis.readthedocs.io
AAanalysis is a Python framework for interpretable, sequence-based protein prediction. It turns sequences into physicochemical features (CPP), trains explainable models, and traces every prediction back to a residue × property × group comparison — robust for small datasets. v1.1 extends the core feature engine beyond physicochemical scales to PLM embeddings and protein structure.
Install · Import · Loadcore + [pro]
# Python >= 3.11 pip install aaanalysis # core pip install 'aaanalysis[pro]' # SHAP, FIMO, Bio import numpy as np import matplotlib.pyplot as plt import aaanalysis as aa df_seq = aa.load_dataset(name='DOM_GSEC') # γ-secretase labels = df_seq['label'].to_list() df_scales = aa.load_scales()
The Golden Workflowcanonical pipeline
1LOADload_dataset · load_scales→df_seq · df_scales
2PARTSSequenceFeature.get_df_parts→df_parts
3FEATURESPart × Split × Scale · CPP.run→df_feat
4MODELTreeModel.fit · dPULearn.fit→feat_importance · labels_
5EXPLAINCPPPlot.feature_map · ShapModel→figure · feat_impact
Prediction Task Levelstask → setup
Residue AA_*positional
unit: sliding window (aa_window_size)
ref: non-site windows / shuffled background
Domain DOM_*both
unit: part-set jmd_n · tmd · jmd_c (from tmd_start / tmd_stop)
ref: labelled A vs B groups
Protein SEQ_*compositional
unit: whole chain (composition)
ref: labelled groups / composition-matched background
Output types: classification · regression · ranking · explanation
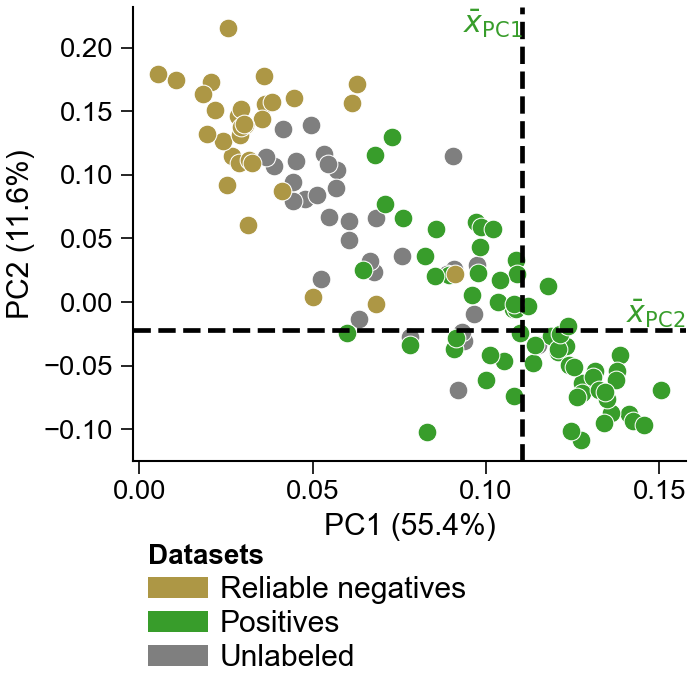
Groups:1: positives0: negatives0: reliable negatives2: unlabeled
Label 0 is a curated negative in labelled data, or a dPULearn-inferred reliable negative drawn from the unlabeled (2) pool.
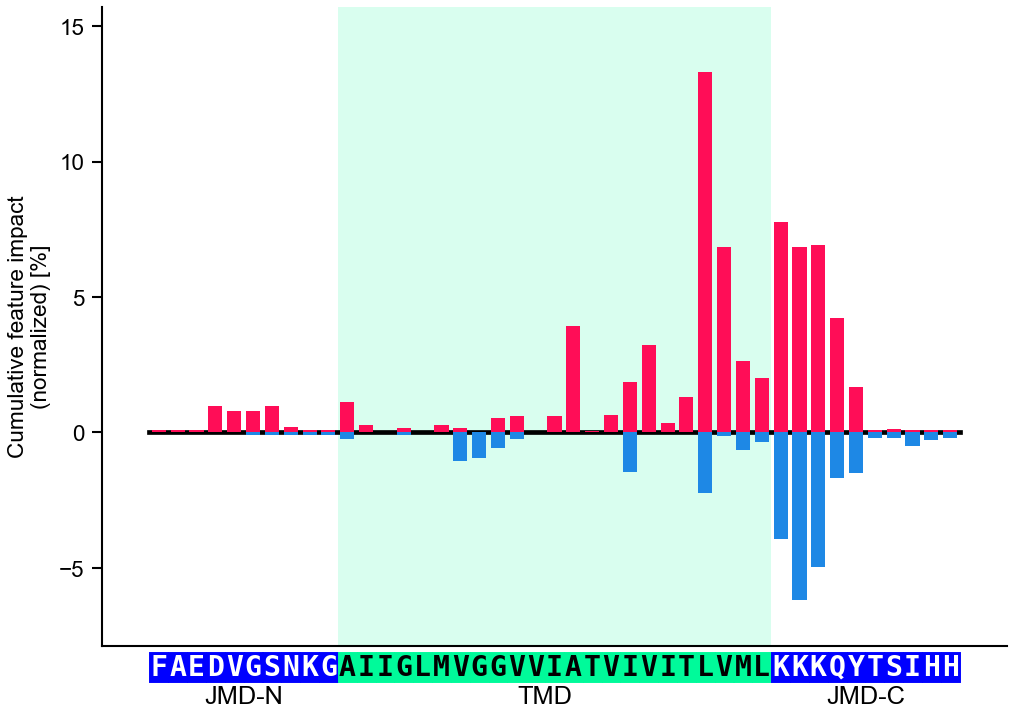
Sequence Anatomythe TMD model
| TMD | Target Middle Domain — the central segment of interest (e.g. transmembrane domain); variable length. |
| JMD | Juxta Middle Domain — the fixed-width flanks adjoining the TMD (jmd_n on the N-side, jmd_c on the C-side). |
JMD-N
TMD
JMD-C
0tmd_starttmd_stoplen(seq)
JMD widths set globally: aa.options['jmd_n_len'] · ['jmd_c_len'].
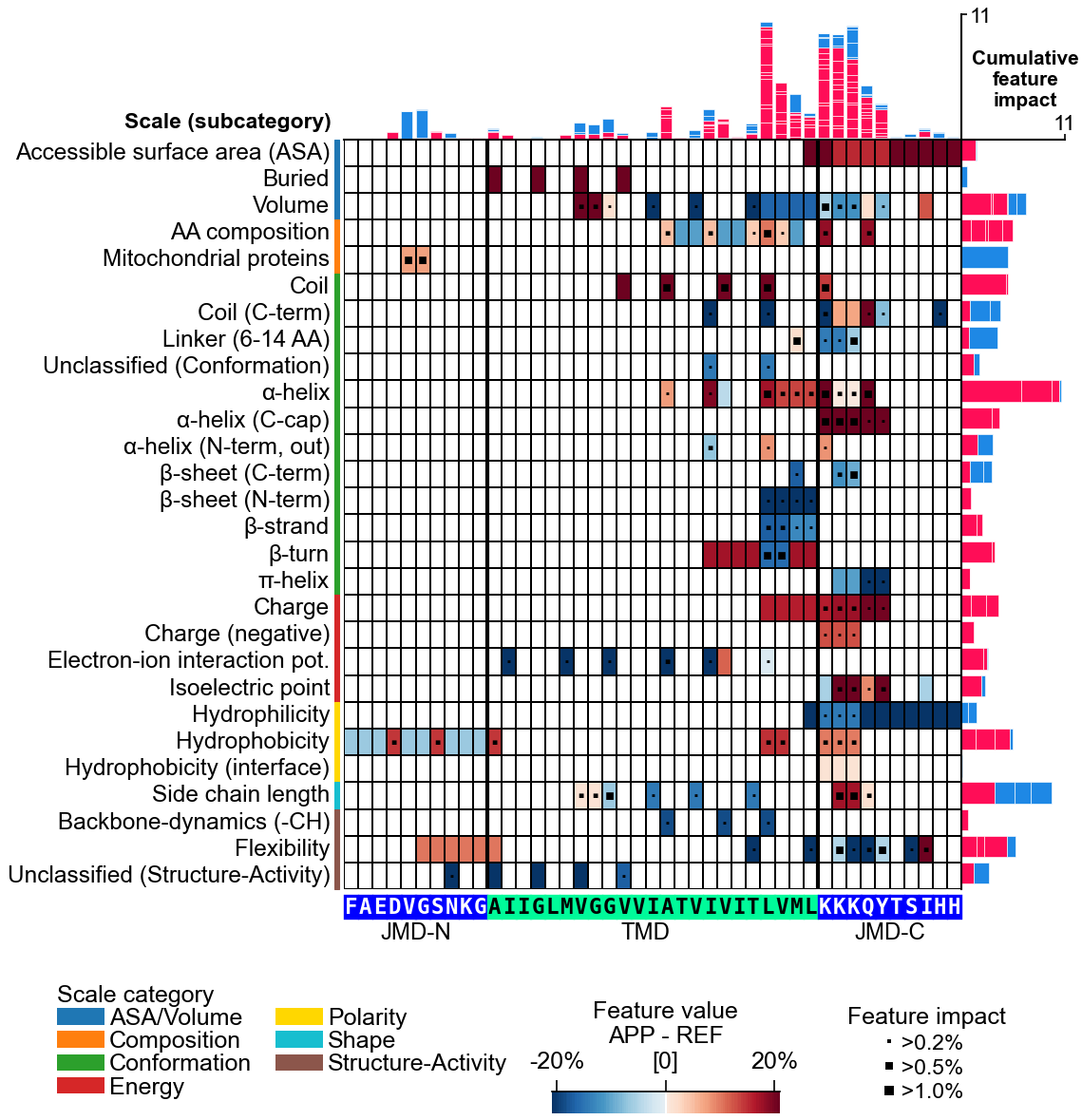
CPP Feature ConceptPart × Split × Scale
PART where on the sequence
tmd · jmd_n · jmd_c · tmd_jmd · jmd_n_tmd_n · tmd_c_jmd_c
SPLIT how to read the part
Segment — contiguous · Pattern — sparse pairs · PeriodicPattern — i, i+3/4
SCALE which physicochemical property
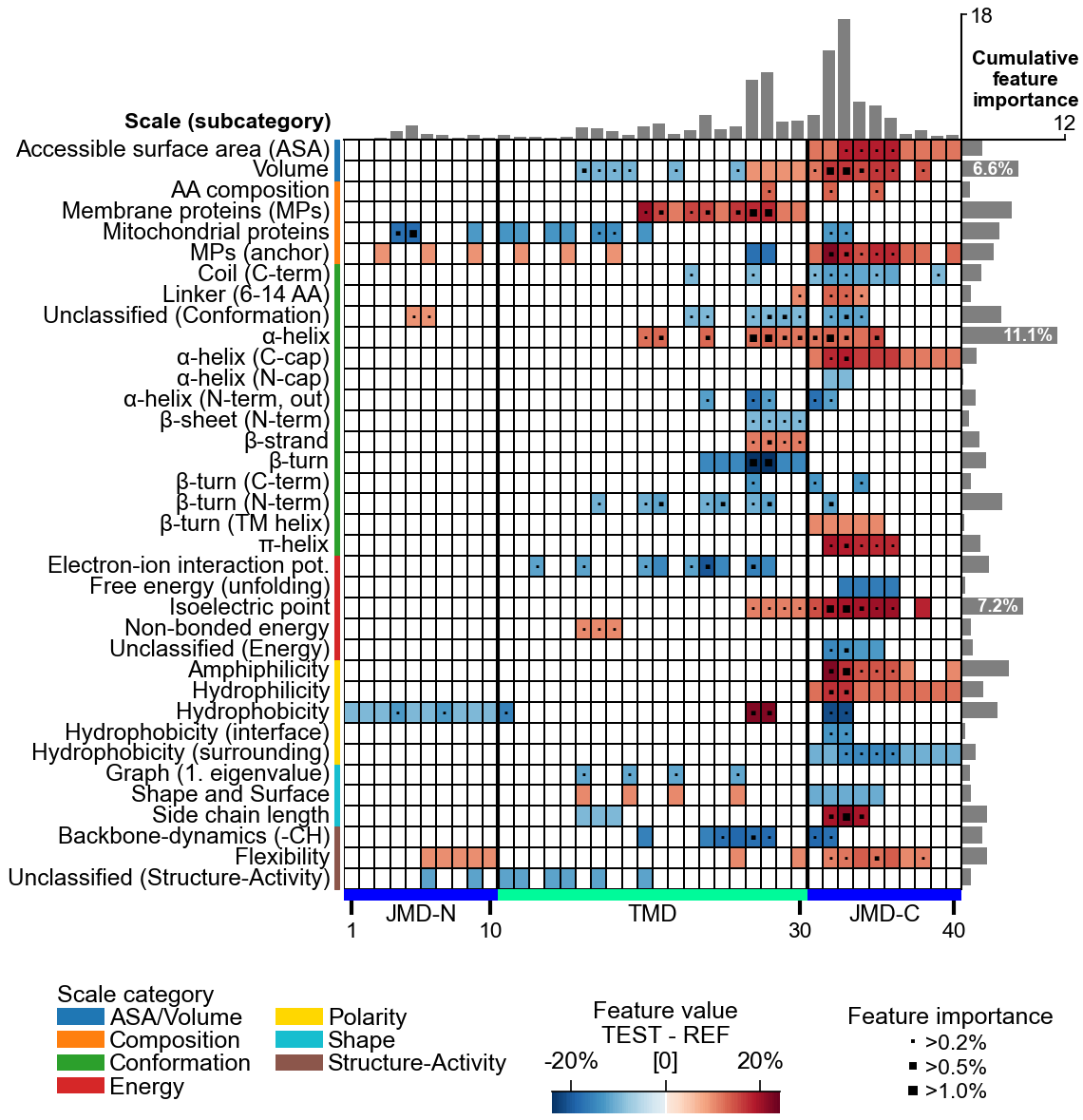
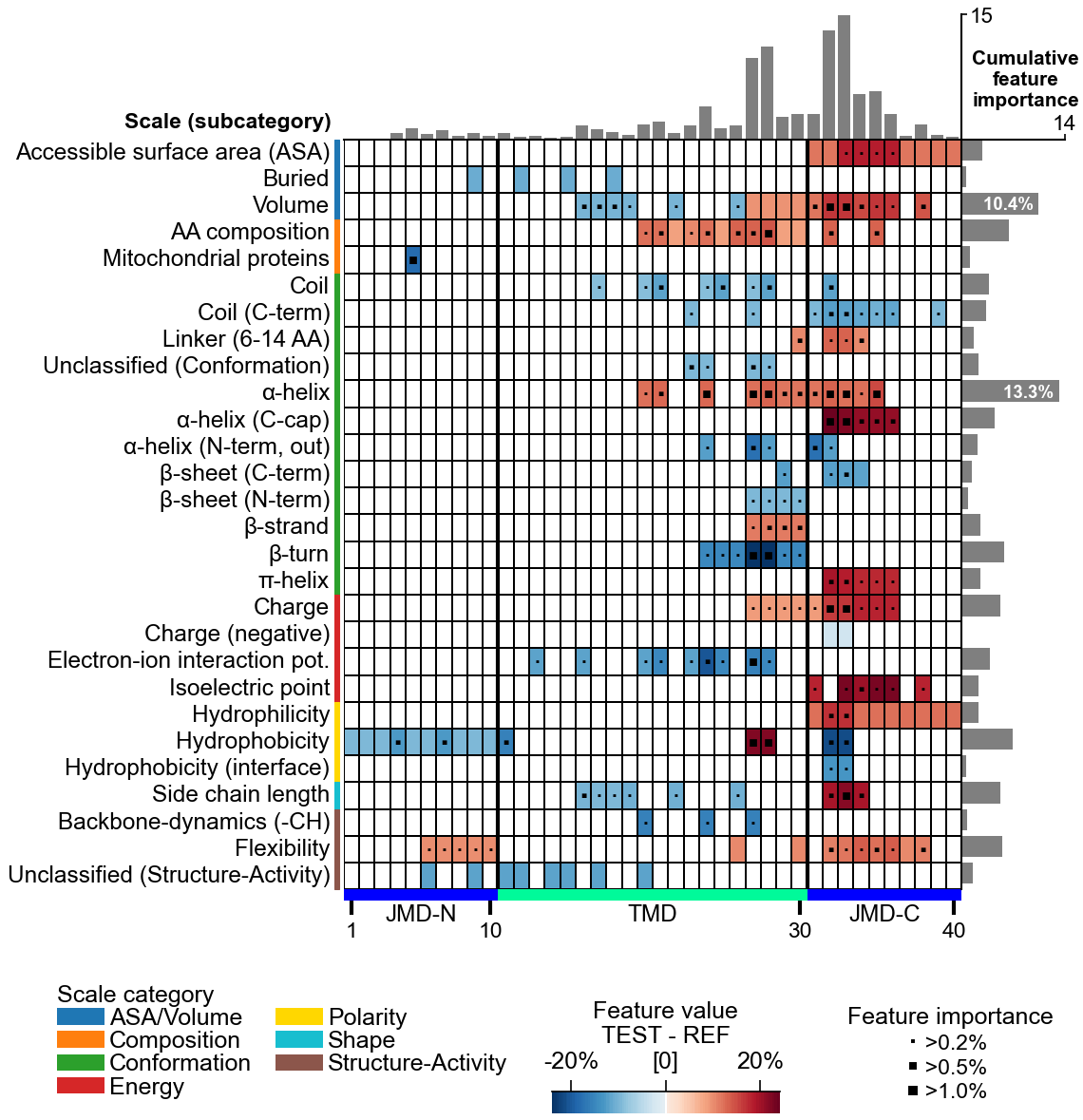
AAontology (~600 scales) · hydrophobicity · charge · helix propensity
TMDA I I G L M V G G V V I
Segment(1,4)■ ■ ■ · · · · · · · · ·
Pattern(N,1,4,8)■ · · ■ · · · ■ · · · ·
PeriodicPattern■ · · ■ · · ■ · · ■ · ·
Splitting maps parts (of various length) to fixed relative positions.
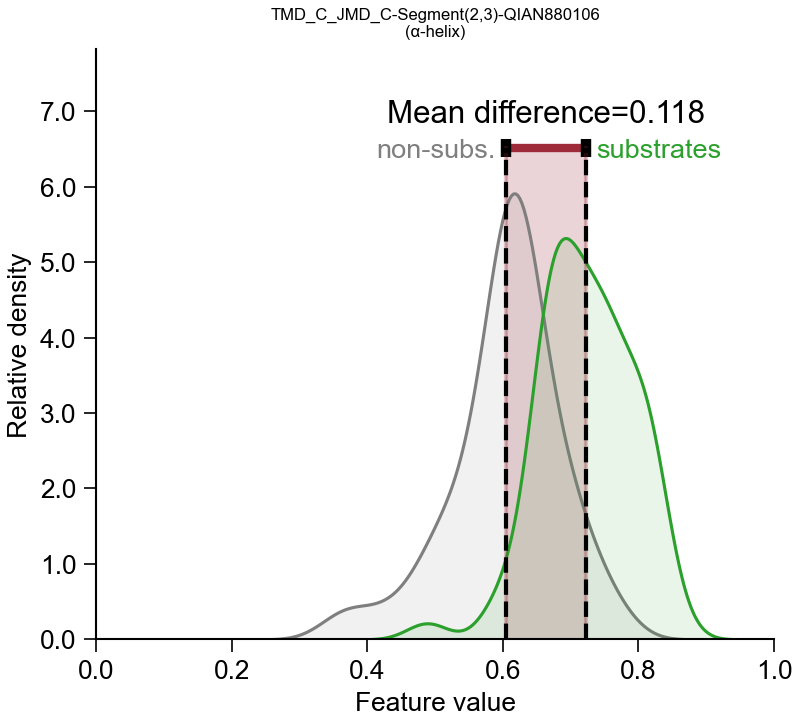
TMD × Segment × hydrophobicity → membrane insertion
JMD × Pattern × net charge → electrostatic recognition
TMD × PeriodicPattern × helix → α-helical interface
CPP Strategiesvia split_kws
Compositional ≈ sequence/protein-level
one whole-part average (composition-like, position-agnostic)
split_kws = sf.get_split_kws(
split_types="Segment",
n_split_max=1)
cpp = aa.CPP(df_parts=df_parts, split_kws=split_kws)
Positional ≈ residue-/region-level
sub-segments and/or patterns resolved to positions
split_kws = sf.get_split_kws(
split_types=["Segment", "Pattern", "PeriodicPattern"],
n_split_max=5,
steps_pattern=[3, 4],
steps_periodicpattern=[3, 4])
cpp = aa.CPP(df_parts=df_parts, split_kws=split_kws)
Domain level uses both. → CPP strategies: see the CPP tutorial (docs).
Which Module Should I Use?intent → module
Explore sequence patterns / compositionAALogo
Sample reference windows (if negatives are missing)AAWindowSampler
Reduce redundant amino acid scalesAAclust
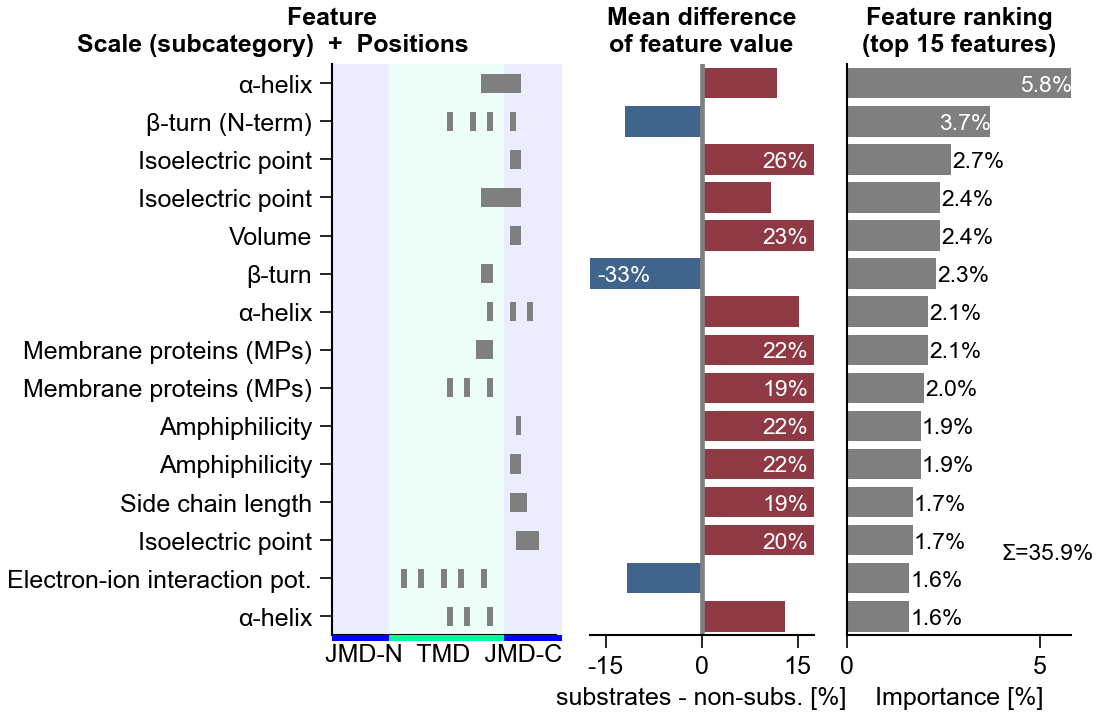
Discover discriminative physicochemical featuresCPP
Train with positives + unlabeled datadPULearn
Train an interpretable classifierTreeModel
Explain a prediction (per feature / sample)ShapModel pro
Data & Preparationdatasets · scales · FASTA
Load benchmark sequencesload_dataset(name) → df_seq
Load AAontology scalesload_scales() → df_scales
Load precomputed featuresload_features(name) → df_feat
Binary labels from df column v1.1get_labels(df, positive_label) → labels
Read / write FASTAread_fasta(file) → df_seq
Cluster redundant homologsfilter_seq(df_seq) → df_clust pro
Sequence Analysislogos · windows · motifs
Position-specific logoAALogo().get_df_logo(df_parts) → df_logo
Sample reference windowsAAWindowSampler().sample_*(df_seq)
Pairwise sequence similaritycomp_seq_sim(df_seq) pro
Scan motifs (FIMO / MEME)scan_motif(df_seq, pwm) → df_hits pro
Feature Engineeringparts · CPP · scales
SequenceFeature → sfsf = aa.SequenceFeature()
· split sequence into partssf.get_df_parts(df_seq) → df_parts
· assemble feature matrix Xsf.feature_matrix(df_feat, df_parts) → X
Discover discriminative featuresCPP(df_parts).run(labels) → df_feat ★
Sweep CPP configs (grid)CPPGrid().run(...) · .eval() → ranked configs
Simplify → interpretable scalesCPP.simplify(df_feat, labels) → df_feat
Reduce redundant scalesAAclust().fit(X) [Wrapper]
Drop correlated featuresNumericalFeature().filter_correlation(X)
Feature Preprocessingone-hot · PLM · structure · PTM
Encode sequences (one-hot / int)SequencePreprocessor().encode_*(seqs) → X
PLM embeddings v1.1EmbeddingPreprocessor().encode(...) → dict_num
Structure / DSSP / PAE v1.1StructurePreprocessor().encode_dssp(...) → dict_num pro
PTM / site annotations v1.1AnnotationPreprocessor().encode(...) → dict_num pro
Combine sources v1.1combine_dict_nums([...]) → dict_num
Numerical CPP v1.1CPP(df_parts).run_num(dict_num_parts, labels) → df_feat
Modeling & ExplainabilityPU · classify · SHAP
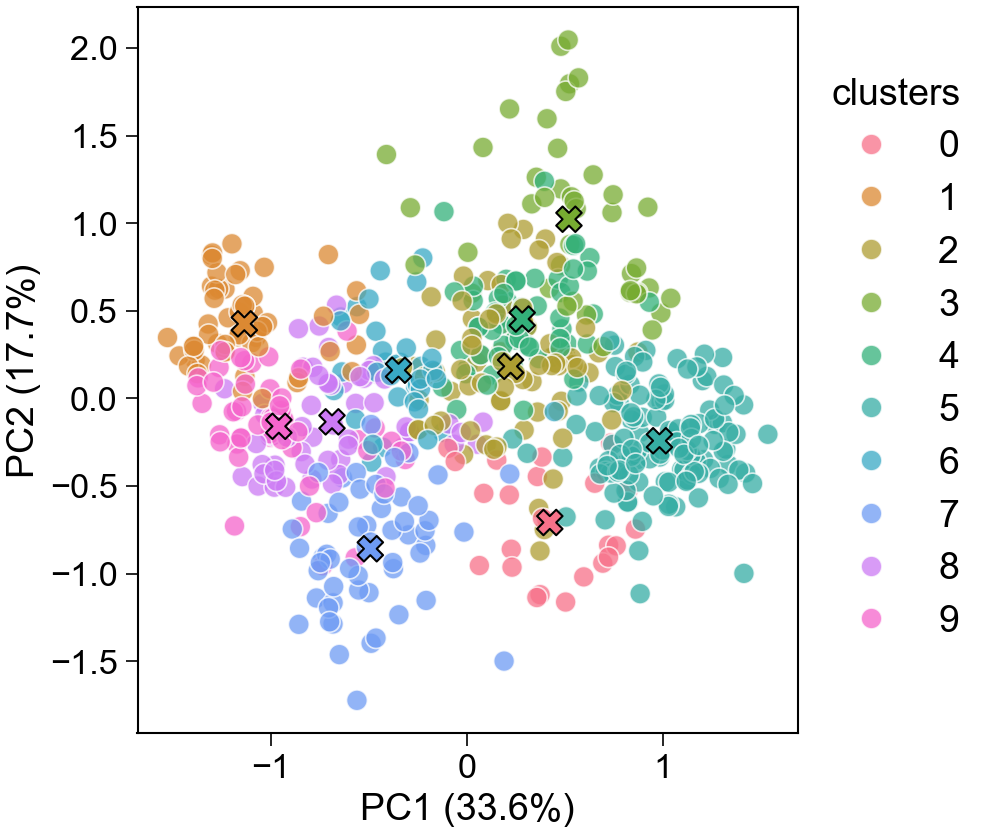
Train with positives + unlabeled datadPULearn().fit(X, labels) [Wrapper]
Mine reliable negatives (mask) v1.1dPULearn().fit(X_pos=, X_unlabeled=).mask_neg_ → mask
Project held-out points into PC space v1.1dPULearn().fit(X, labels).project(X_new) → df_pu
Train + RFE + MC importanceTreeModel().fit(X, labels) [Wrapper]
Per-feature / sample SHAP impactShapModel().fit(X, labels) pro
Metrics & Plottingmetrics · plots
Adjusted AUC (class imbalance)comp_auc_adjusted(X, labels)
BIC score · KL divergencecomp_bic_score(X, labels) · comp_kld
Per-protein / detection (v1.1)comp_per_protein_ap · comp_detection_metrics
Plot style, fonts & standalone legendplot_settings(font_scale) · plot_legend(ax)
Protein Design (to be extended)mutations · design
In-silico point mutations v1.1AAMut · AAMutPlot
Sequence-design libraries v1.1SeqMut · SeqMutPlot