Usage Principles
To get started with AAanalysis, import it as follows:
import aaanalysis as aa
AAanalysis streamlines a Python-based machine learning workflow for protein prediction, starting with protein sequences typically retrieved from UniProt and assessed for similarity by Biopython´s functionalities. It processes redundancy-reduced sets of these sequences to delineate their most discriminative features for machine learning prediction using scikit-learn. For enhanced interpretability, AAanalysis integrates with the SHapley Additive exPlanations (SHAP) framework to provide detailed explanations of prediction results for individual sequences at single-residue resolution.
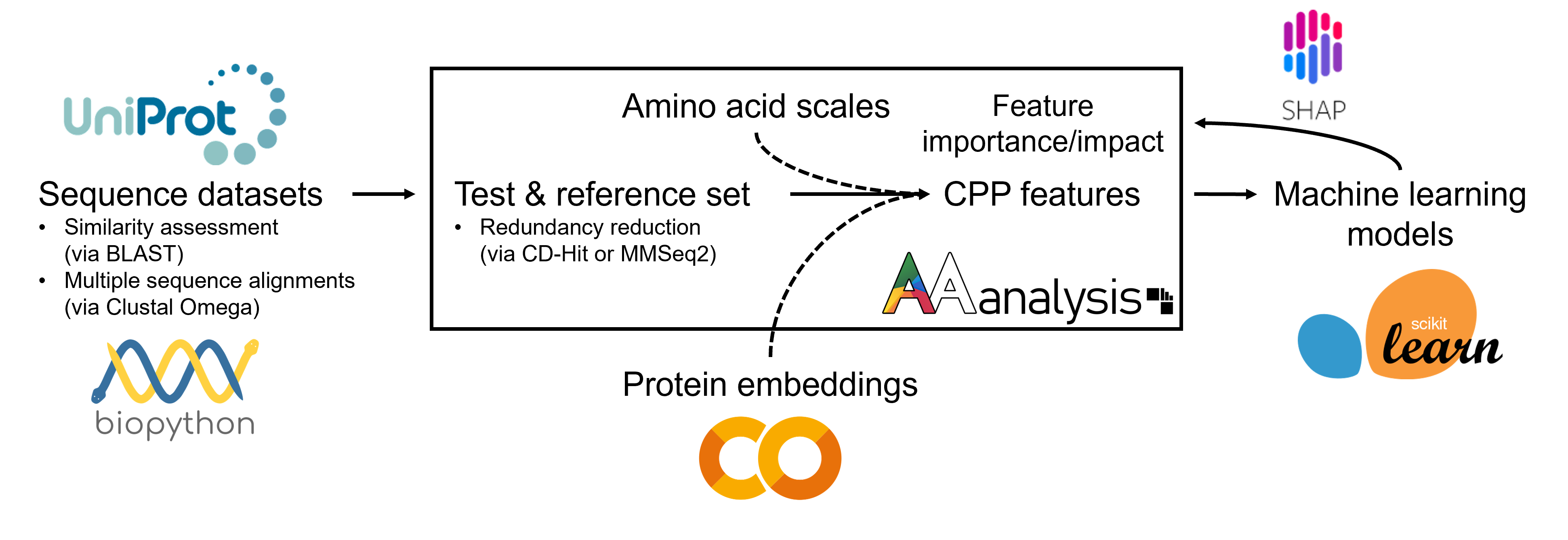
General pipeline for sequence-based protein prediction in Python with AAanalysis. Either amino acid scales or protein embeddings can be used as numerical representation of amino acids (indicated by dashed lines). Protein embeddings can be created via Google Colab and are currently integrated with CPP.
AAanalysis provides a handful of DataFrames for seamless data management. Starting with amino acid scale information (df_scales, df_cat) and protein sequences (df_seq), it enables segmentation into parts (df_parts) and accommodates user-defined splitting (split_kws). Our CPP algorithm then utilizes these to generate physicochemical features (df_feat) by comparing protein sequence sets.
See the primary analysis pipeline of the AAanalysis framework in this diagram:
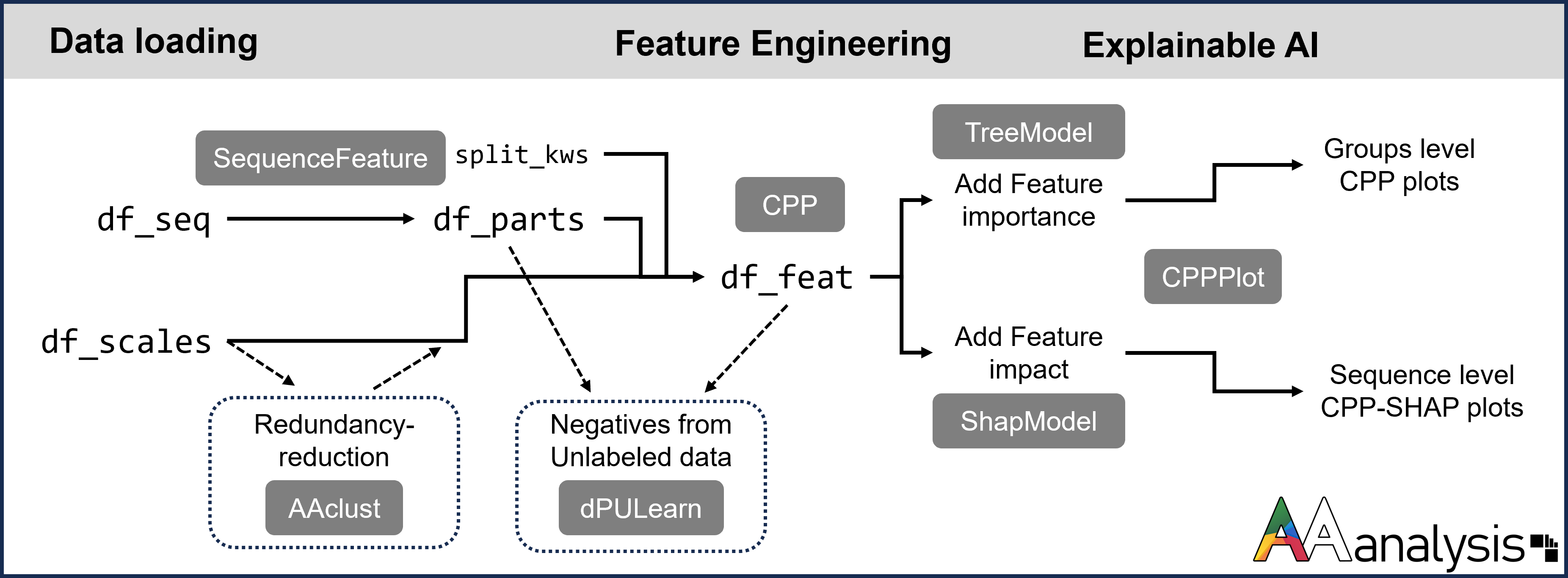
AAanalysis pipeline illustrating the typical data flow, represented as data frames, with key methods (Python classes) highlighted by black squares.
Details on the foundational concepts of AAnalysis are provided by the following sections: