
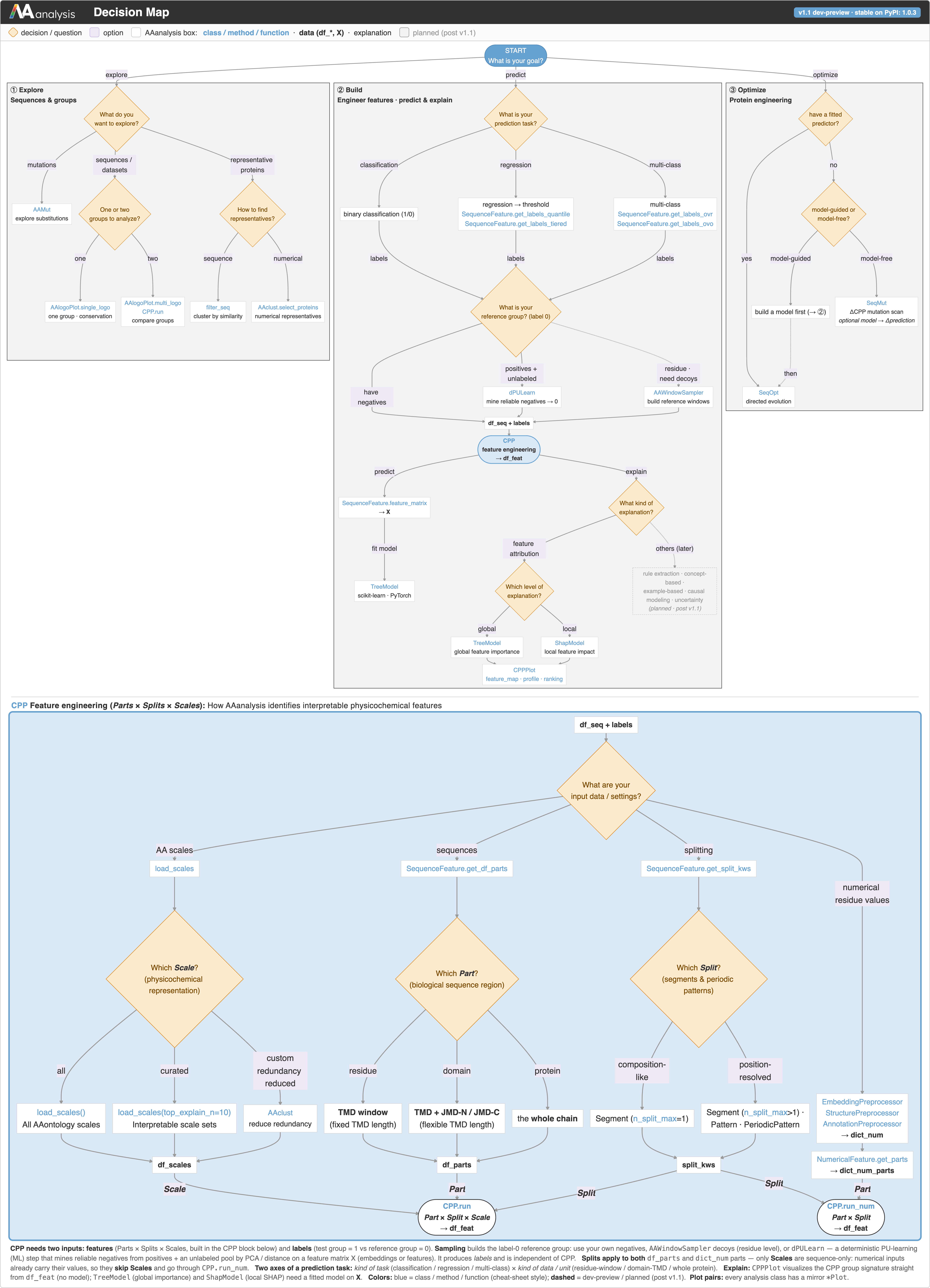
Decision Map
v1.1 dev-preview · stable on PyPI: 1.0.3
decision / question
option
AAanalysis box: class / method / function · data (df_*, X) · explanation
planned (post v1.1)
%%{init: {"flowchart": {"htmlLabels": true, "curve": "basis", "nodeSpacing": 26, "rankSpacing": 30, "padding": 8, "wrappingWidth": 360}}}%%
flowchart TB
START(["START
What is your goal?"]):::start
subgraph EX["① Explore
Sequences & groups"]
QX{"What do you
want to explore?"}:::dec
AAM["AAMut
explore substitutions"]:::fn
Q1{"One or two
groups to analyze?"}:::dec
AAL["AAlogoPlot.single_logo
one group · conservation"]:::fn
AALC["AAlogoPlot.multi_logo
CPP.run
compare groups"]:::fn
QREP{"How to find
representatives?"}:::dec
REPs["filter_seq
cluster by similarity"]:::fn
REPn["AAclust.select_proteins
numerical representatives"]:::fn
end
subgraph PR["② Build
Engineer features · predict & explain"]
SAMP{"What is your
prediction task?"}:::dec
Sc["binary classification (1/0)"]:::rep
Sr["regression → threshold
SequenceFeature.get_labels_quantile
SequenceFeature.get_labels_tiered"]:::rep
Sm["multi-class
SequenceFeature.get_labels_ovr
SequenceFeature.get_labels_ovo"]:::rep
REFQ{"What is your
reference group? (label 0)"}:::dec
AWS["AAWindowSampler
build reference windows"]:::fn
DPU["dPULearn
mine reliable negatives → 0"]:::fn
DSEQ["df_seq + labels"]:::df
FE(["CPP
feature engineering
→ df_feat"]):::core
EXQ{"What kind of
explanation?"}:::dec
EXLVL{"Which level of
explanation?"}:::dec
CPPP["CPPPlot
feature_map · profile · ranking"]:::fn
FM["SequenceFeature.feature_matrix
→ X"]:::fn
MODEL["TreeModel
scikit-learn · PyTorch"]:::fn
TM["TreeModel
global feature importance"]:::fn
SM["ShapModel
local feature impact"]:::fn
XAIP["rule extraction · concept-based ·
example-based · causal modeling · uncertainty
(planned · post v1.1)"]:::plan
end
subgraph EN["③ Optimize
Protein engineering"]
Q4{"have a fitted
predictor?"}:::dec
Q5{"model-guided or
model-free?"}:::dec
SO["SeqOpt
directed evolution"]:::fn
BM["build a model first (→ ②)"]:::rep
SMu["SeqMut
ΔCPP mutation scan
optional model → Δprediction"]:::fn
end
START -->|explore| QX
START -->|predict| SAMP
START -->|optimize| Q4
QX -->|mutations| AAM
QX -->|"sequences / datasets"| Q1
QX -->|"representative proteins"| QREP
Q1 -->|one| AAL
Q1 -->|two| AALC
QREP -->|sequence| REPs
QREP -->|numerical| REPn
SAMP -->|classification| Sc
SAMP -->|regression| Sr
SAMP -->|multi-class| Sm
Sc -->|"labels"| REFQ
Sr -->|"labels"| REFQ
Sm -->|"labels"| REFQ
REFQ -->|"have negatives"| DSEQ
REFQ -->|"positives + unlabeled"| DPU
REFQ -.->|"residue · need decoys"| AWS
DPU --> DSEQ
AWS --> DSEQ
DSEQ --> FE
FE -->|"predict"| FM
FM -->|"fit model"| MODEL
FE -->|"explain"| EXQ
EXQ -->|"feature attribution"| EXLVL
EXQ -.->|"others (later)"| XAIP
EXLVL -->|"global"| TM
EXLVL -->|"local"| SM
TM --> CPPP
SM --> CPPP
Q4 -->|yes| SO
Q4 -->|no| Q5
Q5 -->|"model-guided"| BM
Q5 -->|"model-free"| SMu
BM -.->|then| SO
classDef start fill:#67A1D0,stroke:#3f7bb0,stroke-width:2px,color:#ffffff
classDef dec fill:#fdebcf,stroke:#e0902c,stroke-width:1.8px,color:#7a4a00
classDef core fill:#dbe8f6,stroke:#67A1D0,stroke-width:3.5px,color:#333131,font-weight:bold
classDef fn fill:#ffffff,stroke:#C9C9C9,color:#333131
classDef df fill:#ffffff,stroke:#C9C9C9,color:#333131,font-weight:bold
classDef rep fill:#ffffff,stroke:#C9C9C9,color:#333131
classDef plan fill:#f1f3f5,stroke:#9a9a9a,stroke-dasharray:5 4,color:#9a9a9a
classDef spacer fill:transparent,stroke:transparent,color:transparent
CPP Feature engineering (Parts × Splits × Scales): How AAanalysis identifies interpretable physicochemical features
%%{init: {"flowchart": {"htmlLabels": true, "curve": "basis", "nodeSpacing": 26, "rankSpacing": 30, "padding": 10, "wrappingWidth": 360}}}%%
flowchart TB
%% ---- input router · same df_seq as the main map ----
SEQ["df_seq + labels"]:::df
INQ{"What are your
input data / settings?"}:::dec
SEQ --> INQ
%% ===== column 1 · SCALES (sequence path · the value source) =====
LS["load_scales"]:::fn
SCQ{"Which Scale?
(physicochemical representation)"}:::dec
SALL["load_scales()
All AAontology scales"]:::rep
SSEL["load_scales(top_explain_n=10)
Interpretable scale sets"]:::rep
SAAC["AAclust
reduce redundancy"]:::fn
SCALES["df_scales"]:::df
INQ -->|"AA scales"| LS
LS --> SCQ
SCQ -->|"all"| SALL
SCQ -->|"curated"| SSEL
SCQ -->|"custom
redundancy reduced"| SAAC
SALL --> SCALES
SSEL --> SCALES
SAAC --> SCALES
%% ===== column 2 · PARTS (sequence path) =====
GDP["SequenceFeature.get_df_parts"]:::fn
UNIT{"Which Part?
(biological sequence region)"}:::dec
Uwin["TMD window
(fixed TMD length)"]:::rep
Utmd["TMD + JMD-N / JMD-C
(flexible TMD length)"]:::rep
Uwhole["the whole chain"]:::rep
PARTS["df_parts"]:::df
INQ -->|"sequences"| GDP
GDP --> UNIT
UNIT -->|"residue"| Uwin
UNIT -->|"domain"| Utmd
UNIT -->|"protein"| Uwhole
Uwin --> PARTS
Utmd --> PARTS
Uwhole --> PARTS
%% ===== column 3 · SPLITS (apply to BOTH parts and dict_num parts) =====
GSK["SequenceFeature.get_split_kws"]:::fn
SPQ{"Which Split?
(segments & periodic patterns)"}:::dec
SPC["Segment (n_split_max=1)"]:::rep
SPP["Segment (n_split_max>1) ·
Pattern · PeriodicPattern"]:::rep
SPLITS["split_kws"]:::df
INQ -->|"splitting"| GSK
GSK --> SPQ
SPQ -->|"composition-like"| SPC
SPQ -->|"position-resolved"| SPP
SPC --> SPLITS
SPP --> SPLITS
%% ===== column 4 · numerical values (alternative to SCALES; only feeds run_num) =====
PREP["EmbeddingPreprocessor
StructurePreprocessor
AnnotationPreprocessor
→ dict_num"]:::fn
NFP["NumericalFeature.get_parts
→ dict_num_parts"]:::fn
INQ -->|"numerical residue values"| PREP
PREP --> NFP
%% ---- CPP cores ----
CPP([" CPP.run
Part × Split × Scale
→ df_feat"]):::algo
CPPN([" CPP.run_num
Part × Split
→ df_feat"]):::algo
SCALES -->|"Scale"| CPP
PARTS -->|"Part"| CPP
SPLITS -->|"Split"| CPP
NFP -->|"Part"| CPPN
SPLITS -->|"Split"| CPPN
classDef dec fill:#fdebcf,stroke:#e0902c,stroke-width:1.8px,color:#7a4a00
classDef algo fill:#ffffff,stroke:#333131,stroke-width:2.5px,color:#333131,font-weight:bold
classDef fn fill:#ffffff,stroke:#C9C9C9,color:#333131
classDef df fill:#ffffff,stroke:#C9C9C9,color:#333131,font-weight:bold
classDef rep fill:#ffffff,stroke:#C9C9C9,color:#333131
classDef spacer fill:transparent,stroke:transparent,color:transparent
CPP needs two inputs: features (Parts × Splits × Scales, built in the CPP block below) and labels (test group = 1 vs reference group = 0). Sampling builds the label-0 reference group: use your own negatives,
AAWindowSampler decoys (residue level), or dPULearn — a deterministic PU-learning (ML) step that mines reliable negatives from positives + an unlabeled pool by PCA / distance on a feature matrix X (embeddings or features). It produces labels and is independent of CPP.
Splits apply to both df_parts and dict_num parts — only Scales are sequence-only: numerical inputs already carry their values, so they skip Scales and go through CPP.run_num.
Two axes of a prediction task: kind of task (classification / regression / multi-class) × kind of data / unit (residue-window / domain-TMD / whole protein).
Explain: CPPPlot visualizes the CPP group signature straight from df_feat (no model); TreeModel (global importance) and ShapModel (local SHAP) need a fitted model on X.
Colors: blue = class / method / function (cheat-sheet style); dashed = dev-preview / planned (post v1.1). Plot pairs: every analysis class has a mirror *Plot.
{kind=link}