Slow start with AAanalysis
Dive into the powerful capabilities of AAanalysis—a Python framework for sequence-based (i.e., alignment-free) and interpretable protein prediction. In this tutorial, we’ll focus on extracting physicochemical features from protein sequences using the Comparative Physical Profiling (CPP) algorithm and how these features can be harnessed for binary classification tasks.
What You Will Learn
Data Loadings: Load protein sequences and selections of amino acid scales.
Feature Engineering: Extract essential features using the
AAclustandCPPmodels.Machine Learning: Make predictions using the RandomForest model.
Explainable AI: Interpret predictions at the group and individual levels by combining
CPPwithSHAP.
import seaborn as sns
import matplotlib.pyplot as plt
import pandas as pd
import numpy as np
import aaanalysis as aa
aa.options["verbose"] = False
aa.options["random_state"] = 42
1. Data Loading
With AAanalysis, you have access to numerous benchmark datasets for
sequence-based protein prediction, mainly for binary classification
tasks. You can retrieve these datasets with the aa.load_dataset()
function. Amino acid scales, predominantly from
AAindex, along with their two-level
hierarchical classification (AAontology, [Breimann24b]), are
available with the aa.load_scales() function. See AAontology Usage
Principels
for further details.
We first load the complete scales dataset and our primary example dataset from [Breimann25a]_ of γ-secretase substrates (n=63) and non-substrates (n=63):
# Load example dataset and scales
df_seq = aa.load_dataset(name="DOM_GSEC")
labels = list(df_seq["label"])
df_scales = aa.load_scales()
2. Feature Engineering
The centerpiece of AAanalysis is the CPP model, which is supported
by AAclust, a clustering wrapper framework for the pre-selection of
amino acid scales, which was introduced in [Breimann24a].
AAclust
Since redundancy is an essential problem for machine learning tasks, the
AAclust object provides a lightweight wrapper for sklearn clustering
algorithms such as Agglomerative clustering. AAclust clusters a set of
scales and selects for each cluster the most representative scale (i.e.,
the scale closes to the cluster center). More details can be found in
AAclust Usage
Principels.
We will use AAclust to obtain a set of 100 scales, as defined by the
n_clusters parameters:
# Obtain redundancy-reduced set of 100 scales
aac = aa.AAclust()
X = np.array(df_scales).T
scales = aac.fit(X, names=list(df_scales), n_clusters=100).medoid_names_
df_scales = df_scales[scales]
Comparative Physicochemical Profiling (CPP)
CPP is a sequence-based algorithm for interpretable feature engineering, introduced in [Breimann25a]. It aims at identifying a set of features most discriminant between two sets of sequences: the test set and the reference set. The core idea of CPP is its feature concepts, which integrates:
Parts: Are combination of a target middle domain (TMD) and N- and C-terminal adjacent regions (JMD-N and JMD-C, respectively), obtained
sf.get_df_parts.Splits: These Parts can be split into various continuous segments or discontinuous patterns, specified
sf.get_split_kws().Scales: Sets of amino acid scales (or other numerical scales).
See CPP Usage Principles for more conceptual background.
We use the supporting SequenceFeature class to obtain Parts and
Splits. These together with the Scales are used by CPP as input
to identify the set of characteristic features to discriminate between
both datasets, such as γ-secretase substrates and non-substrates:
sf = aa.SequenceFeature()
# Obtain Parts and Splits
df_parts = sf.get_df_parts(df_seq=df_seq, list_parts=["tmd"])
split_kws = sf.get_split_kws(n_split_max=1, split_types=["Segment"])
Performing the CPP algorithm using the CPP.run() method creates all
Part-Split-Split combinations and filters them down to a
user-defined maximum of non-redundant features (100 by default). As
baseline, we use CPP without filtering (max_cor=1) to compute the
average values for the 100 selected scales over the entire TMD
sequences:
# Create 100 baseline features (Scale values averaged over TMD)
cpp = aa.CPP(df_scales=df_scales, df_parts=df_parts, split_kws=split_kws)
df_feat_baseline = cpp.run(labels=labels, max_cor=1)
aa.display_df(df=df_feat_baseline, n_rows=8)
| feature | category | subcategory | scale_name | scale_description | abs_auc | abs_mean_dif | mean_dif | std_test | std_ref | p_val_mann_whitney | p_val_fdr_bh | positions | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TMD-Segment(1,1)-WOLR790101 | Polarity | Hydrophobicity (surrounding) | Hydration potential | Hydrophobicity ...n et al., 1979) | 0.257000 | 0.037000 | 0.037000 | 0.028000 | 0.042000 | 0.000001 | 0.000064 | 11,12,13,14,15,...,26,27,28,29,30 |
| 2 | TMD-Segment(1,1)-FUKS010106 | Composition | Membrane proteins (MPs) | Proteins of mesophiles (INT) | Interior compos...ishikawa, 2001) | 0.240000 | 0.068000 | 0.068000 | 0.065000 | 0.082000 | 0.000003 | 0.000160 | 11,12,13,14,15,...,26,27,28,29,30 |
| 3 | TMD-Segment(1,1)-BEGF750103 | Conformation | β-turn | β-turn | Conformational ...in-Dirkx, 1975) | 0.237000 | 0.064000 | -0.064000 | 0.067000 | 0.087000 | 0.000004 | 0.000147 | 11,12,13,14,15,...,26,27,28,29,30 |
| 4 | TMD-Segment(1,1)-FASG760105 | Polarity | Unclassified (Polarity) | pK-C | pK-C (Fasman, 1976) | 0.234000 | 0.044000 | 0.044000 | 0.038000 | 0.057000 | 0.000006 | 0.000142 | 11,12,13,14,15,...,26,27,28,29,30 |
| 5 | TMD-Segment(1,1)-YUTK870104 | Energy | Free energy (unfolding) | Free energy (unfolding) | Activation Gibb...i et al., 1987) | 0.234000 | 0.011000 | 0.011000 | 0.010000 | 0.020000 | 0.000006 | 0.000121 | 11,12,13,14,15,...,26,27,28,29,30 |
| 6 | TMD-Segment(1,1)-LINS030116 | ASA/Volume | Accessible surface area (ASA) | ASA (folded β-strand) | Total median ac...s et al., 2003) | 0.230000 | 0.028000 | -0.028000 | 0.021000 | 0.039000 | 0.000008 | 0.000137 | 11,12,13,14,15,...,26,27,28,29,30 |
| 7 | TMD-Segment(1,1)-ANDN920101 | Structure-Activity | Backbone-dynamics (-CH) | α-CH chemical s...kbone-dynamics) | alpha-CH chemic...n et al., 1992) | 0.229000 | 0.068000 | -0.068000 | 0.073000 | 0.081000 | 0.000009 | 0.000102 | 11,12,13,14,15,...,26,27,28,29,30 |
| 8 | TMD-Segment(1,1)-ROBB760109 | Conformation | β-turn (N-term) | β-turn (1st residue) | Information mea...n-Suzuki, 1976) | 0.229000 | 0.039000 | -0.039000 | 0.041000 | 0.047000 | 0.000009 | 0.000115 | 11,12,13,14,15,...,26,27,28,29,30 |
CPP uses 330 Splits and 3 Parts by default, yielding around 100,000 features when using 100 Scales. Creating and filtering all these Part-Split-Scale combinations will take a little time (about 25 seconds for this example on an i7-10510U (4 cores, 8 threads) with multiprocessing), but it significantly improves prediction performance:
# CPP creates around 100,000 features and filters them down to 100
df_parts = sf.get_df_parts(df_seq=df_seq)
cpp = aa.CPP(df_scales=df_scales, df_parts=df_parts)
df_feat = cpp.run(labels=labels)
aa.display_df(df=df_feat, n_rows=8)
| feature | category | subcategory | scale_name | scale_description | abs_auc | abs_mean_dif | mean_dif | std_test | std_ref | p_val_mann_whitney | p_val_fdr_bh | positions | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...2,3)-QIAN880106 | Conformation | α-helix | α-helix (middle) | Weights for alp...ejnowski, 1988) | 0.387000 | 0.118000 | 0.118000 | 0.068000 | 0.080000 | 0.000000 | 0.000000 | 27,28,29,30,31,32,33 |
| 2 | TMD_C_JMD_C-Pat...,12)-ROBB760109 | Conformation | β-turn (N-term) | β-turn (1st residue) | Information mea...n-Suzuki, 1976) | 0.363000 | 0.119000 | -0.119000 | 0.065000 | 0.085000 | 0.000000 | 0.000000 | 21,25,28,32 |
| 3 | TMD_C_JMD_C-Seg...6,9)-ZIMJ680104 | Energy | Isoelectric point | Isoelectric point | Isoelectric poi...n et al., 1968) | 0.352000 | 0.268000 | 0.268000 | 0.174000 | 0.173000 | 0.000000 | 0.000000 | 32,33 |
| 4 | TMD_C_JMD_C-Seg...4,9)-ROBB760113 | Conformation | β-turn | β-turn | Information mea...n-Suzuki, 1976) | 0.349000 | 0.337000 | -0.337000 | 0.177000 | 0.254000 | 0.000000 | 0.000000 | 27,28 |
| 5 | TMD_C_JMD_C-Seg...4,5)-ZIMJ680104 | Energy | Isoelectric point | Isoelectric point | Isoelectric poi...n et al., 1968) | 0.336000 | 0.205000 | 0.205000 | 0.134000 | 0.157000 | 0.000000 | 0.000000 | 33,34,35,36 |
| 6 | TMD_C_JMD_C-Pat...,15)-QIAN880106 | Conformation | α-helix | α-helix (middle) | Weights for alp...ejnowski, 1988) | 0.336000 | 0.152000 | 0.152000 | 0.102000 | 0.132000 | 0.000000 | 0.000000 | 28,32,35 |
| 7 | TMD_C_JMD_C-Seg...6,9)-LINS030101 | ASA/Volume | Volume | Accessible surface area (ASA) | Total accessibl...s et al., 2003) | 0.328000 | 0.235000 | 0.235000 | 0.156000 | 0.187000 | 0.000000 | 0.000000 | 32,33 |
| 8 | TMD_C_JMD_C-Seg...2,3)-ZIMJ680104 | Energy | Isoelectric point | Isoelectric point | Isoelectric poi...n et al., 1968) | 0.326000 | 0.108000 | 0.108000 | 0.077000 | 0.085000 | 0.000000 | 0.000000 | 27,28,29,30,31,32,33 |
3. Machine Learning (Protein Prediction)
A feature matrix for a given set of CPP features can be created using
the sf.feat_matrix() method. The feature matrix will be used to
train a Random Forest
Classifier
model:
from sklearn.ensemble import RandomForestClassifier
from sklearn.model_selection import cross_val_score
# Create feature matrix and perform prediction for baseline features
X = sf.feature_matrix(df_parts=df_parts, features=df_feat_baseline["feature"])
rf = RandomForestClassifier()
cv_base = cross_val_score(rf, X, labels, scoring="accuracy", cv=5)
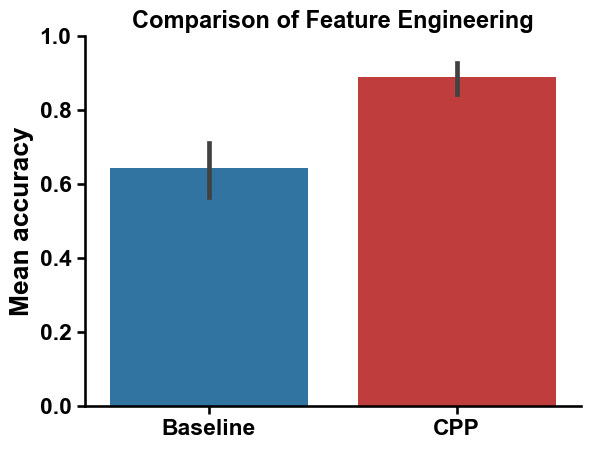
print(f"Mean accuracy of {round(np.mean(cv_base), 2)}")
Mean accuracy of 0.64
We create now the feature matrix for the features obtained with CPP default settings:
# Create feature matrix and perform prediction for default CPP features
X = sf.feature_matrix(df_parts=df_parts, features=df_feat["feature"])
rf = RandomForestClassifier()
cv = cross_val_score(rf, X, labels, scoring="accuracy", cv=5)
print(f"Mean accuracy of {round(np.mean(cv), 2)}")
Mean accuracy of 0.89
We can compare the baseline and default CPP feature set using a bar chart:
# Comparison of baseline and default CPP feature sets
aa.plot_settings()
sns.barplot(pd.DataFrame({"Baseline": cv_base, "CPP": cv}), palette=["tab:blue", "tab:red"])
plt.ylabel("Mean accuracy", size=aa.plot_gcfs()+1)
plt.ylim(0, 1)
plt.title("Comparison of Feature Engineering", size=aa.plot_gcfs()-1)
sns.despine()
plt.show()
CPP Analysis
AAanalysis offers a range of plotting functions through the CPPPlot
class for interpreting the features identified by CPP. For a binary
classification tasks, the CPP features and their machine learning-based
importance can be visualized at two different levels:
Group level: Highlights the differences and feature importance between two groups, aiding in their discrimination.
Sample level: Shows differences between an individual sample and a reference group, along with the importance of specific features.
CPP Analysis (group level)
To obtain the group level feature importance, we developed the
TreeModel class. It is a wrapper for tree-based models from
scikit-learn or similar libraries
XGBoost or
CatBoost. The TreeModel obtains a
Monte Carlo estimate of the importance of each feature by training
different models over multiple iterations and averaging their results.
These estimates can be included into the feature DataFrame (df_feat)
using the TreeModel.add_feat_importance() method:
# Include group level feature importance
tm = aa.TreeModel()
tm.fit(X, labels=labels)
df_feat = tm.add_feat_importance(df_feat=df_feat)
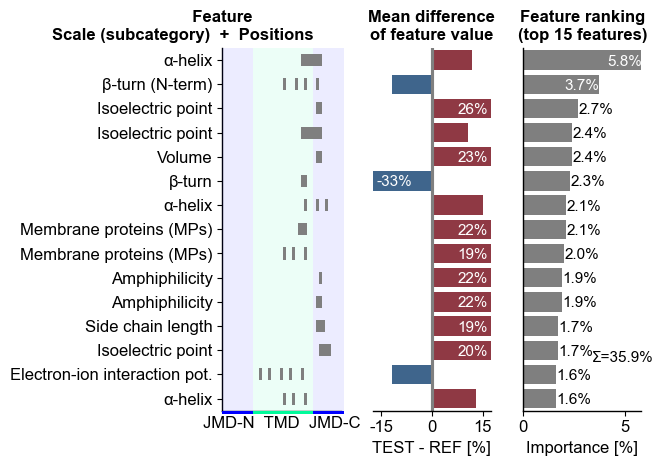
The top15 features can be displayed using the CPPPlot.ranking()
method. The features are shown (left) as a combination of the scale
subcategory and their position derived from the Part-Split
combination. The difference of feature values between the test and
reference set of protein (here γ-secretase substrates and
non-substrates) is shown in the middle. The right subplots displays the
feature importance used for ranking:
# Plot CPP ranking
cpp_plot = aa.CPPPlot()
aa.plot_settings(short_ticks=True, weight_bold=False)
cpp_plot.ranking(df_feat=df_feat)
plt.tight_layout()
plt.show()
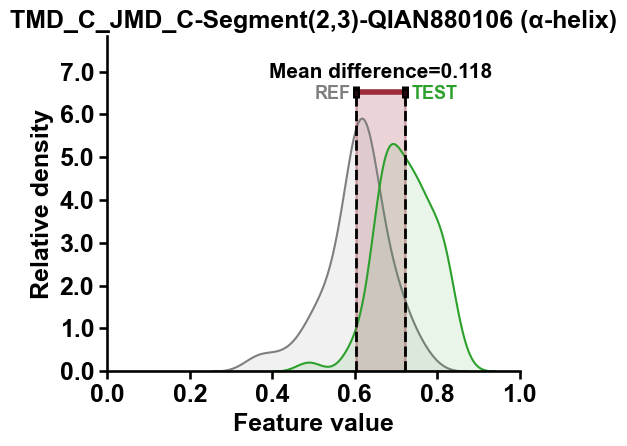
The distributions of features values for the highest-ranked feature can
be displayed using the CPPPlot.feature() method, which shows the
distribution for the test (‘Test’) and reference (‘REF’) dataset along
with the difference of their mean values (‘Mean difference’):
top_feature = df_feat["feature"][0]
top_subcategory = df_feat["subcategory"][0]
aa.plot_settings()
cpp_plot.feature(feature=top_feature , df_seq=df_seq, labels=labels)
plt.title(f"{top_feature} ({top_subcategory})")
plt.tight_layout()
plt.show()
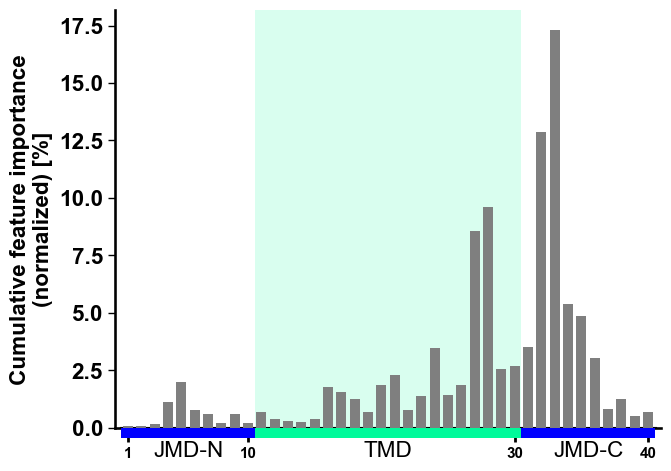
To visualize the importance of all features at single-residue
resolution, the cumulative feature importance per residue position can
be shown using the CPPPlot.profile() method. As this method presents
group level results, it allows for the adjustment of the lengths of
different Parts:
# Plot CPP profile
aa.plot_settings(font_scale=0.9)
cpp_plot.profile(df_feat=df_feat)
plt.tight_layout()
plt.show()
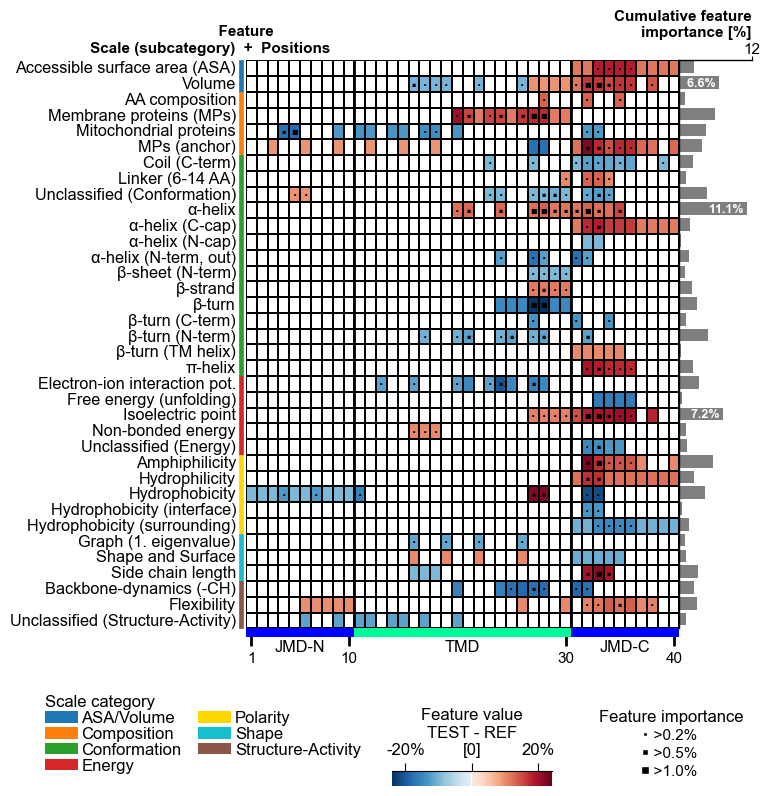
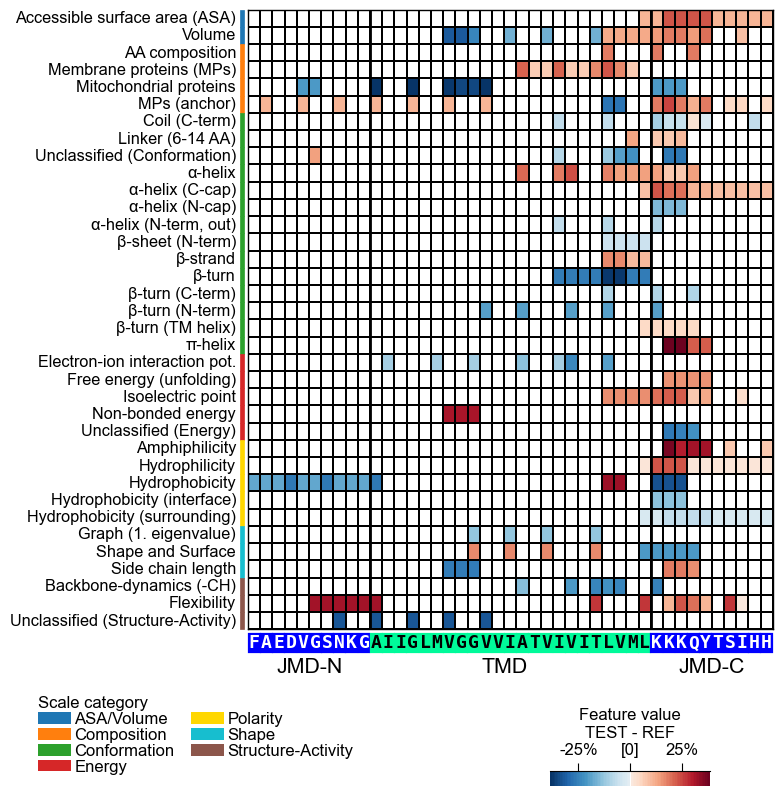
The centerpiece of the AAanalysis package is the feature map, which shows the common physicochemical signature for discriminating the test from the reference protein set. It is created by the `CPPPlot.feature_map()`` method showing the feature value differences per scale subcategory at single-residue resolution:
# Plot CPP feature map (original version: without importance bars on top)
cpp_plot = aa.CPPPlot()
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.feature_map(df_feat=df_feat, add_imp_bar_top=False)
plt.show()
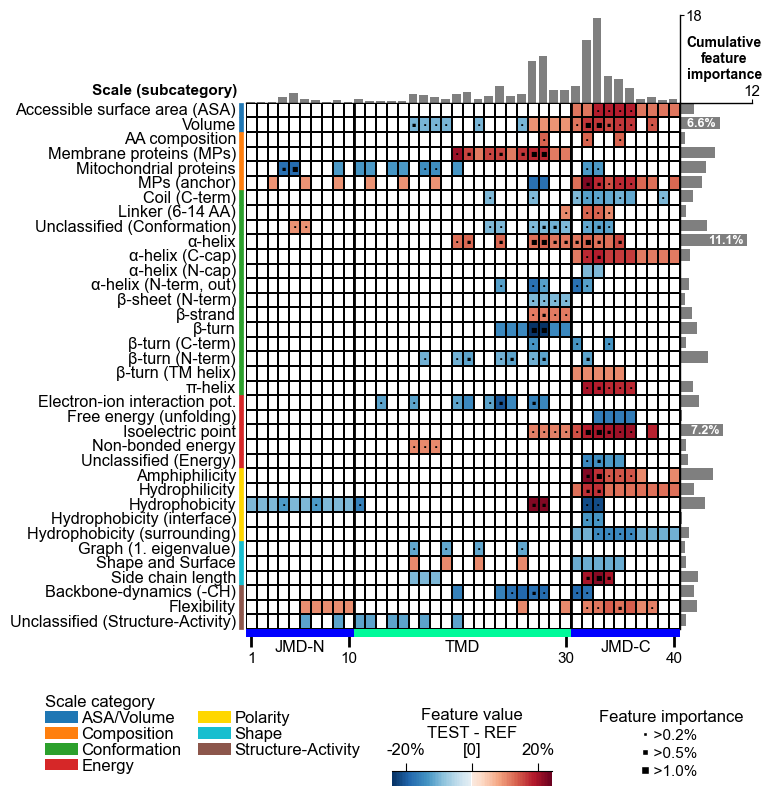
# Plot CPP feature map (v1.0.2+: with importance bars on top)
cpp_plot = aa.CPPPlot()
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.feature_map(df_feat=df_feat)
plt.show()
4. Explainable AI
CPP-SHAP Analysis (sample level)
We can analyse the impact of features on the prediction score for
individual sequence using the ShapModel model, which combines
CPP with the explainable AI framework
SHAP.
# Obtain sample-specific feature impact
sm = aa.ShapModel()
sm.fit(X, labels=labels)
df_feat = sm.add_feat_impact(df_feat=df_feat)
df_feat = sm.add_sample_mean_dif(X, labels=labels, df_feat=df_feat)
To explain the feature impact with single-residue resolution, AAanalysis offers the following four types of visualizations: CPP-SHAP ranking plot, CPP-SHAP profile, CPP heatmap, CPP-SHAP heatmap. An overview is provided under Explainable AI Usage Principles
We can now show the feature ranking for a selected protein (‘Protein0’)
using the CPPPlot.ranking() method, plotting SHAP analysis results
by setting shap_plot=True:
# CPP-SHAP ranking plot
aa.plot_settings(short_ticks=True, weight_bold=False)
cpp_plot.ranking(df_feat=df_feat, shap_plot=True, col_dif="mean_dif_Protein0", col_imp="feat_impact_Protein0")
plt.tight_layout()
plt.show()
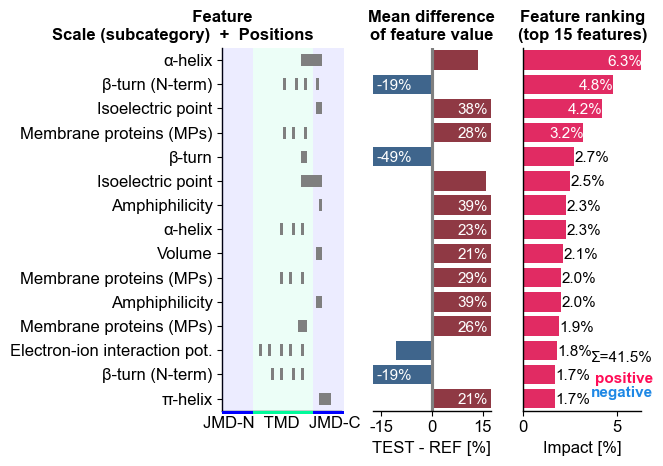
To visualize the CPP-SHAP profile and heatmap, we need to obtain the sequence parts of the respective protein:
# Get sequences parts for APP
_df_parts = sf.get_df_parts(df_seq=df_seq, list_parts=["tmd", "jmd_c", "jmd_n"])
_args_seq = _df_parts.loc["P05067"].to_dict()
args_seq = {key + "_seq": _args_seq[key] for key in _args_seq}
Show the specific CPP-SHAP Profile for the first Protein using the
CPPPlot.profile() method with setting shap_plot=True:
# CPP-SHAP profile
aa.plot_settings(font_scale=0.9)
cpp_plot.profile(df_feat=df_feat, shap_plot=True, col_imp="feat_impact_Protein0", **args_seq)
plt.tight_layout()
plt.show()
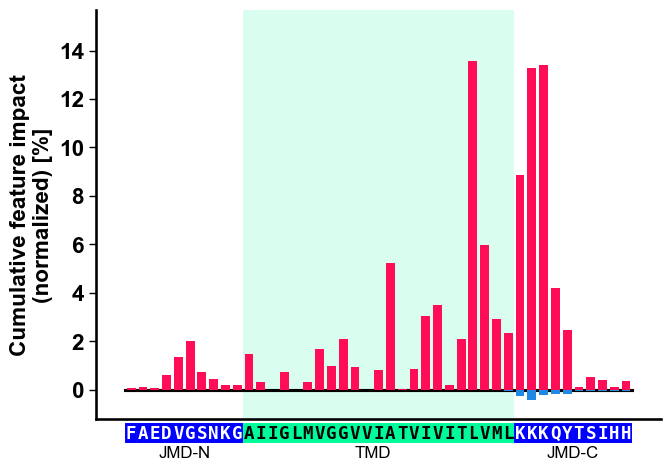
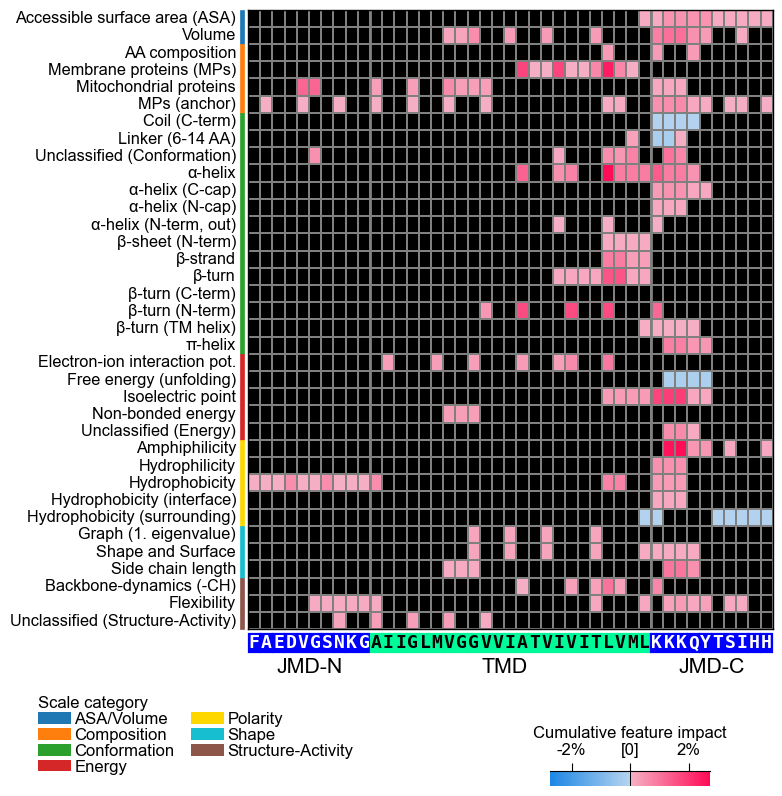
With the CPPPlot.heatmap() method we can visualize the
sample-specific feature value difference and the feature impact per
scale subcategory and residue position. Set shap_plot=True and
provide the respective column with the mean difference and the feature
impact:
# CPP heatmap (sample level)
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.heatmap(df_feat=df_feat, shap_plot=True, col_val="mean_dif_Protein0", **args_seq)
plt.show()
# CPP-SHAP heatmap (sample level)
cpp_plot.heatmap(df_feat=df_feat, shap_plot=True, col_val="feat_impact_Protein0", **args_seq)
plt.show()
See our Feature Engineering
API
for a comprehensive documentation on the CPP, CPPPlot,
AAclust, and SequenceFeature classes.