Data loading
This is a tutorial on loading of protein benchmark datasets.
You will learn
Tool:
load_dataset()Input: dataset
name(+n,min_len,max_len,aa_window_size)Output:
df_seq(sequence table with the binarylabelcolumn)Best used for: loading labelled protein benchmark datasets at amino-acid, domain, or sequence level
Related protocol: P4: Prediction levels
Related API:
load_dataset(),load_features()
Loading of protein benchmarks
Load the overview table of protein benchmark datasets using the default settings:
import aaanalysis as aa
df_info = aa.load_dataset()
aa.display_df(df=df_info)
| Level | Dataset | # Sequences | Avg length | # Amino acids | # Positives | # Negatives | Predictor | Description | Reference | Label | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Amino acid | AA_CASPASE3 | 233 | 796.587983 | 185605 | 705 | 184900 | PROSPERous | Prediction of c...3 cleavage site | Song et al., 2018 | 1 (adjacent to ... cleavage site) |
| 2 | Amino acid | AA_FURIN | 71 | 831.028169 | 59003 | 163 | 58840 | PROSPERous | Prediction of f...n cleavage site | Song et al., 2018 | 1 (adjacent to ... cleavage site) |
| 3 | Amino acid | AA_LDR | 342 | 345.754386 | 118248 | 35469 | 82779 | IDP-Seq2Seq | Prediction of l...d regions (LDR) | Tang et al., 2020 | 1 (disordered), 0 (ordered) |
| 4 | Amino acid | AA_MMP2 | 573 | 546.205934 | 312976 | 2416 | 310560 | PROSPERous | Prediction of M...) cleavage site | Song et al., 2018 | 1 (adjacent to ... cleavage site) |
| 5 | Amino acid | AA_RNABIND | 221 | 248.873303 | 55001 | 6492 | 48509 | GMKSVM-RU | Prediction of R...(RBP60 dataset) | Yang et al., 2021 | 1 (binding), 0 (non-binding) |
| 6 | Amino acid | AA_SA | 233 | 796.587983 | 185605 | 101082 | 84523 | PROSPERous | Prediction of s...PASE3 data set) | Song et al., 2018 | 1 (exposed/acce...non-accessible) |
| 7 | Sequence | SEQ_AMYLO | 1414 | 6.000000 | 8484 | 511 | 903 | ReRF-Pred | Prediction of a...ognenic regions | Teng et al. 2021 | 1 (amyloidogeni...-amyloidogenic) |
| 8 | Sequence | SEQ_CAPSID | 7935 | 424.030246 | 3364680 | 3864 | 4071 | VIRALpro | Prediction of capdsid proteins | Galiez et al., 2016 | 1 (capsid prote...capsid protein) |
| 9 | Sequence | SEQ_DISULFIDE | 2547 | 241.252454 | 614470 | 897 | 1650 | Dipro | Prediction of d...es in sequences | Cheng et al., 2006 | 1 (sequence wit...ithout SS bond) |
| 10 | Sequence | SEQ_LOCATION | 1835 | 399.126975 | 732398 | 1045 | 790 | nan | Prediction of s...lasma membrane) | Shen et al., 2019 | 1 (protein in c...asma membrane) |
| 11 | Sequence | SEQ_SOLUBLE | 17408 | 254.611041 | 4432269 | 8704 | 8704 | SOLpro | Prediction of s...oluble proteins | Magnan et al., 2009 | 1 (soluble), 0 (insoluble) |
| 12 | Sequence | SEQ_TAIL | 6668 | 400.673365 | 2671690 | 2574 | 4094 | VIRALpro | Prediction of tail proteins | Galiez et al., 2016 | 1 (tail protein...n-tail protein) |
| 13 | Domain | DOM_GSEC | 126 | 737.809524 | 92964 | 63 | 63 | nan | Prediction of g...tase substrates | Breimann et al, 2024c | 1 (substrate), ...(non-substrate) |
| 14 | Domain | DOM_GSEC_PU | 694 | 712.570605 | 494524 | 63 | 0 | nan | Prediction of g...es (PU dataset) | Breimann et al, 2024c | 1 (substrate), ...bstrate status) |
The benchmark datasets are categorized into amino acid (‘AA’), domain
(‘DOM’), and sequence (‘SEQ’) level datasets, indicated by their
name prefix, as exemplified here.
df_seq1 = aa.load_dataset(name="AA_CASPASE3")
df_seq2 = aa.load_dataset(name="SEQ_CAPSID")
df_seq3 = aa.load_dataset(name="DOM_GSEC")
aa.display_df(df=df_seq3, n_rows=8, char_limit=25)
# Compare columns of three types
| entry | sequence | label | tmd_start | tmd_stop | jmd_n | tmd | jmd_c | |
|---|---|---|---|---|---|---|---|---|
| 1 | P05067 | MLPGLALLLLAA...NPTYKFFEQMQN | 1 | 701 | 723 | FAEDVGSNKG | AIIGLMVGGVVIATVIVITLVML | KKKQYTSIHH |
| 2 | P14925 | MAGRARSGLLLL...YSAPLPKPAPSS | 1 | 868 | 890 | KLSTEPGSGV | SVVLITTLLVIPVLVLLAIVMFI | RWKKSRAFGD |
| 3 | P70180 | MRSLLLFTFSAC...REDSIRSHFSVA | 1 | 477 | 499 | PCKSSGGLEE | SAVTGIVVGALLGAGLLMAFYFF | RKKYRITIER |
| 4 | Q03157 | MGPTSPAARGQG...ENPTYRFLEERP | 1 | 585 | 607 | APSGTGVSRE | ALSGLLIMGAGGGSLIVLSLLLL | RKKKPYGTIS |
| 5 | Q06481 | MAATGTAAAAAT...NPTYKYLEQMQI | 1 | 694 | 716 | LREDFSLSSS | ALIGLLVIAVAIATVIVISLVML | RKRQYGTISH |
| 6 | P35613 | MAAALFVLLGFA...DKGKNVRQRNSS | 1 | 323 | 345 | IITLRVRSHL | AALWPFLGIVAEVLVLVTIIFIY | EKRRKPEDVL |
| 7 | P35070 | MDRAARCSGASS...PINEDIEETNIA | 1 | 119 | 141 | LFYLRGDRGQ | ILVICLIAVMVVFIILVIGVCTC | CHPLRKRRKR |
| 8 | P09803 | MGARCRSFSALL...KLADMYGGGEDD | 1 | 711 | 733 | GIVAAGLQVP | AILGILGGILALLILILLLLLFL | RRRTVVKEPL |
Each dataset can be utilized for a binary classification, with labels
being positive (1) or negative (0). A balanced number of samples can be
chosen by the n parameter, defining the sample number per class.
# Returns 200 samples, the first 100 positives and the first 100 negatives
df_seq = aa.load_dataset(name="SEQ_CAPSID", n=100)
aa.display_df(df=df_seq, n_rows=8, char_limit=25)
| entry | sequence | label | |
|---|---|---|---|
| 1 | CAPSID_1 | MVTHNVKINKHV...RIPATKLDEENV | 0 |
| 2 | CAPSID_2 | MKKRQKKMTLSN...EAVINARHFGEE | 0 |
| 3 | CAPSID_3 | MRYGGSVISQEL...GAPDKQEVELVD | 0 |
| 4 | CAPSID_4 | MERGDIPFKYVG...LEELAEMDAGLI | 0 |
| 5 | CAPSID_5 | MKRIYLLFAALI...CVGEPITELTHQ | 0 |
| 6 | CAPSID_6 | MARARESQAEAL...RNTISSRISRTW | 0 |
| 7 | CAPSID_8 | MLFKQLFTIISS...VDIIKALIIIKG | 0 |
| 8 | CAPSID_9 | MKILIVEDEPKT...GMGYVLEAPQPR | 0 |
Or randomly selected using random=True:
# Returns randoms samples, 100 positives and 100 negatives
df_seq = aa.load_dataset(name="SEQ_CAPSID", n=100, random=True)
aa.display_df(df=df_seq, n_rows=8, char_limit=25)
| entry | sequence | label | |
|---|---|---|---|
| 1 | CAPSID_135 | MIRLLIADDQAL...LAAETLTSGKAH | 0 |
| 2 | CAPSID_540 | MKIFSVGIENFR...PEPEDDFADLLG | 0 |
| 3 | CAPSID_2406 | MAIFGTGGIAAM...VEGNAPTSKAAK | 0 |
| 4 | CAPSID_2846 | MFSASTTPEQPL...ETRDIMPIDWSV | 0 |
| 5 | CAPSID_429 | MALSPAVAPLRT...TALRALITGQPA | 0 |
| 6 | CAPSID_754 | MTRPTRVVIVDD...EHGVLQGTGVNV | 0 |
| 7 | CAPSID_2093 | MSISAAQIKNLK...NGGGGGSSGGWL | 0 |
| 8 | CAPSID_2140 | MAASGGEGSRDV...SALRDPNVVIVY | 0 |
The protein sequences can have varying length:
# Plot distribution
import matplotlib.pyplot as plt
import seaborn as sns
# Utility AAanalysis function for publication ready plots
aa.plot_settings(font_scale=1.2)
df_seq = aa.load_dataset(name="SEQ_CAPSID", n=100)
list_seq_lens = df_seq["sequence"].apply(len)
sns.histplot(list_seq_lens, binwidth=50)
sns.despine()
plt.xlim(0, 1500)
plt.show()
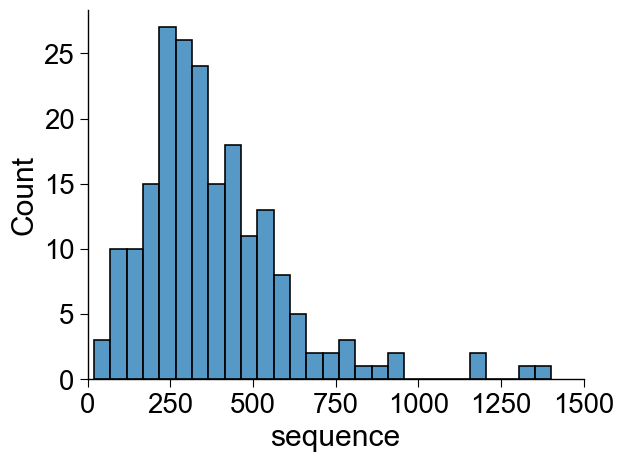
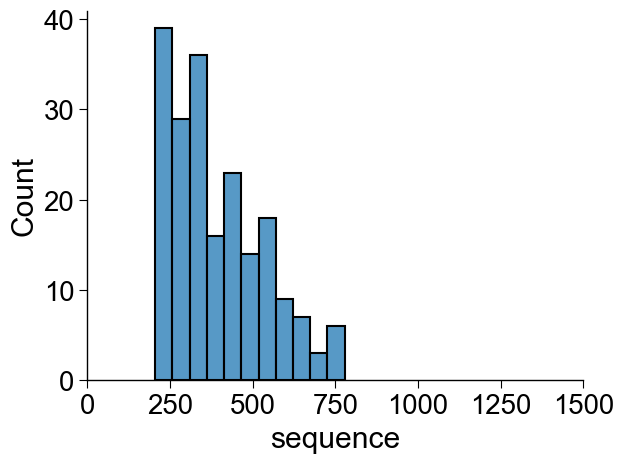
Which can be easily filtered using min_len and max_len
parameters:
df_seq = aa.load_dataset(name="SEQ_CAPSID", n=100, min_len=200, max_len=800)
list_seq_lens = df_seq["sequence"].apply(len)
aa.plot_settings(font_scale=1.2) # Utility AAanalysis function for publication ready plots
sns.histplot(list_seq_lens, binwidth=50)
sns.despine()
plt.xlim(0, 1500)
plt.show()
Loading of protein benchmarks: Amino acid window size
For amino acid level datasets, labels are provided for each residue
position, which can be seen by setting aa_window_size=None:
df_seq = aa.load_dataset(name="AA_CASPASE3", aa_window_size=None)
aa.display_df(df=df_seq.head(10), char_limit=25)
| entry | sequence | label | |
|---|---|---|---|
| 1 | CASPASE3_1 | MSLFDLFRGFFG...LDLFLGRWFRSR | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 2 | CASPASE3_2 | MEVTGDAGVPES...LQNPKRARQDPT | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 3 | CASPASE3_3 | MRARSGARGALL...EMLVAMTTDGDC | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 4 | CASPASE3_4 | MDAKARNCLLQH...NLGILYILQTLE | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 5 | CASPASE3_5 | MTSFSTSAQCST...KEIQLVIKVFIA | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 6 | CASPASE3_6 | MGLGASSEQPAG...PDPEPGLCEGPW | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 7 | CASPASE3_7 | MANQVNGNAVQL...EFYQDTYGQQWK | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 8 | CASPASE3_8 | MAKQPSDVSSEC...LRYIVRLVWRMH | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 9 | CASPASE3_9 | MCTALSPKVRSG...VSASYKAKKEIK | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
| 10 | CASPASE3_10 | MFYAHFVLSKRG...IIATPGPRFHII | 0,0,0,0,0,0,...,0,0,0,0,0,0 |
For convenience, we provide an “amino acid window” of length n. This window represents a specific amino acid, which is flanked by (n-1)/2 residues on both its N-terminal and C-terminal sides. It’s essential for n to be odd, ensuring equal residues on both sides. While the default window size is 9, sizes between 5 and 15 are also popular.
df_seq = aa.load_dataset(name="AA_CASPASE3", n=2)
aa.display_df(df=df_seq, char_limit=25)
| entry | sequence | label | |
|---|---|---|---|
| 1 | CASPASE3_1_pos4 | MSLFDLFRG | 0 |
| 2 | CASPASE3_1_pos5 | SLFDLFRGF | 0 |
| 3 | CASPASE3_1_pos126 | QTLRDSMLK | 1 |
| 4 | CASPASE3_1_pos127 | TLRDSMLKY | 1 |
Sequences can be pre-filtered using min_len and max_len and
n residues can be randomly selected by random with different
aa_window_sizes.
df_seq = aa.load_dataset(name="AA_CASPASE3", min_len=20, n=2, random=True, aa_window_size=21)
aa.display_df(df=df_seq, char_limit=25)
| entry | sequence | label | |
|---|---|---|---|
| 1 | CASPASE3_69_pos39 | EVGTVMTLFYSKKSQRPERKT | 0 |
| 2 | CASPASE3_228_pos316 | EGEEIPRVKPEEMMDERPKTR | 0 |
| 3 | CASPASE3_91_pos119 | LKKQEEEEMDFRSGSPSDNSG | 1 |
| 4 | CASPASE3_179_pos163 | VEVAGDLEVDCYRAPCSESQE | 1 |
Loading of protein benchmarks: Positive-Unlabeled (PU) datasets
In typical binary classification, data is labeled as positive (1) or
negative (0). But with many protein sequence datasets, we face
challenges: they might be small, unbalanced, or lack a clear negative
class. For datasets with only positive and unlabeled samples (2), we use
PU learning. This approach identifies reliable negatives from the
unlabeled data to make binary classification possible. We offer
benchmark datasets for this scenario, denoted by the _PU suffix. For
example, the DOM_GSEC_PU dataset corresponds to the
DOM_GSEC set.
df_seq = aa.load_dataset(name="DOM_GSEC", n=2)
aa.display_df(df=df_seq, char_limit=25)
| entry | sequence | label | tmd_start | tmd_stop | jmd_n | tmd | jmd_c | |
|---|---|---|---|---|---|---|---|---|
| 1 | Q14802 | MQKVTLGLLVFL...TPPLITPGSAQS | 0 | 37 | 59 | NSPFYYDWHS | LQVGGLICAGVLCAMGIIIVMSA | KCKCKFGQKS |
| 2 | Q86UE4 | MAARSWQDELAQ...QIKKKKKARRET | 0 | 50 | 72 | LGLEPKRYPG | WVILVGTGALGLLLLFLLGYGWA | AACAGARKKR |
| 3 | P05067 | MLPGLALLLLAA...NPTYKFFEQMQN | 1 | 701 | 723 | FAEDVGSNKG | AIIGLMVGGVVIATVIVITLVML | KKKQYTSIHH |
| 4 | P14925 | MAGRARSGLLLL...YSAPLPKPAPSS | 1 | 868 | 890 | KLSTEPGSGV | SVVLITTLLVIPVLVLLAIVMFI | RWKKSRAFGD |
df_seq_pu = aa.load_dataset(name="DOM_GSEC_PU", n=2)
aa.display_df(df=df_seq_pu, char_limit=25)
| entry | sequence | label | tmd_start | tmd_stop | jmd_n | tmd | jmd_c | |
|---|---|---|---|---|---|---|---|---|
| 1 | P05067 | MLPGLALLLLAA...NPTYKFFEQMQN | 1 | 701 | 723 | FAEDVGSNKG | AIIGLMVGGVVIATVIVITLVML | KKKQYTSIHH |
| 2 | P14925 | MAGRARSGLLLL...YSAPLPKPAPSS | 1 | 868 | 890 | KLSTEPGSGV | SVVLITTLLVIPVLVLLAIVMFI | RWKKSRAFGD |
| 3 | P12821 | MGAASGRRGPGL...PQFGSEVELRHS | 2 | 1257 | 1276 | GLDLDAQQAR | VGQWLLLFLGIALLVATLGL | SQRLFSIRHR |
| 4 | P36896 | MAESAGASSFFP...LSQLSVQEDVKI | 2 | 127 | 149 | EHPSMWGPVE | LVGIIAGPVFLLFLIIIIVFLVI | NYHQRVYHNR |
See out Data Handling API and the Tables section for more details.