SeqOpt: Optimizing sequences by directed evolution
Protein engineering vs. de novo protein design are two distinct paradigms. De novo design builds new proteins from the ground up rather than repurposing an existing one, typically via a structure-first pipeline — RFdiffusion (backbone) → ProteinMPNN (sequence) → AlphaFold (validation) (Yang et al., 2026). Protein engineering instead optimizes an existing protein by iterative mutation and selection (directed evolution); the machine-learning-guided flavour learns a fitness model to prioritize which variants to make (Yang et al., 2019; Wittmann et al., 2021).
SeqOpt (pro) is that ML-guided directed evolution as a
multi-objective search: it mutates one wild-type and returns the
Pareto front of variants (NSGA-II; a model-bound SeqMut is the
fitness engine), read with SeqOptPlot. Here we engineer a “super
substrate” for gamma-secretase (GSEC): take a non-substrate
transmembrane domain and mutate it to maximize the predicted substrate
probability with as few mutations as possible.
import numpy as np, pandas as pd
import matplotlib.pyplot as plt
from sklearn.ensemble import RandomForestClassifier
import aaanalysis as aa
aa.options["verbose"] = False
aa.plot_settings()
1. Data and a substrate classifier
Load the gamma-secretase dataset and the bundled interpretable CPP feature set, build the feature matrix, and train a simple RandomForest substrate classifier.
df_feat = aa.load_features(name="DOM_GSEC")
df_seq = aa.load_dataset(name="DOM_GSEC", n=50)
labels = df_seq["label"].to_list() # 1 = GSEC substrate
aa.display_df(df_seq, n_rows=5, show_shape=True)
DataFrame shape: (100, 8)
| entry | sequence | label | tmd_start | tmd_stop | jmd_n | tmd | jmd_c | |
|---|---|---|---|---|---|---|---|---|
| 1 | Q14802 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 0 | 37 | 59 | NSPFYYDWHS | LQVGGLICAGVLCAMGIIIVMSA | KCKCKFGQKS |
| 2 | Q86UE4 | MAARSWQDELAQQAE...SPKQIKKKKKARRET | 0 | 50 | 72 | LGLEPKRYPG | WVILVGTGALGLLLLFLLGYGWA | AACAGARKKR |
| 3 | Q969W9 | MHRLMGVNSTAAAAA...AIWSKEKDKQKGHPL | 0 | 41 | 63 | FQSMEITELE | FVQIIIIVVVMMVMVVVITCLLS | HYKLSARSFI |
| 4 | P53801 | MAPGVARGPTPYWRL...GLFKEENPYARFENN | 0 | 97 | 119 | RWGVCWVNFE | ALIITMSVVGGTLLLGIAICCCC | CCRRKRSRKP |
| 5 | Q8IUW5 | MAPRALPGSAVLAAA...EVPATPVKRERSGTE | 0 | 59 | 81 | NDTGNGHPEY | IAYALVPVFFIMGLFGVLICHLL | KKKGYRCTTE |
sf = aa.SequenceFeature()
X = np.asarray(sf.feature_matrix(features=df_feat["feature"],
df_parts=sf.get_df_parts(df_seq=df_seq),
df_scales=aa.load_scales()), dtype=float)
model = RandomForestClassifier(n_estimators=100, random_state=0).fit(X, labels)
print("training accuracy:", round(model.score(X, labels), 3))
training accuracy: 1.0
/Users/stephanbreimann/Programming/1Packages/wt-seqopt-deap/aaanalysis/feature_engineering/_backend/cpp_run.py:163: UserWarning: CPP is using the Python kernel fallback — the compiled Cython extension is not available in this install. Output is bit-exact with the Cython path but ~2x slower. Reinstall via pip install --force-reinstall aaanalysis to fetch a prebuilt wheel. warnings.warn(
2. The design task
Pick a non-substrate as the wild-type. Two objectives: maximize the
predicted substrate probability shift (delta_pred) and minimize the
number of mutations (n_mut).
wt = df_seq[df_seq["label"] == 0].iloc[[0]].reset_index(drop=True)
p_wt = model.predict_proba(X[df_seq["label"].values == 0][:1])[0, 1]
print("wild-type:", wt["entry"].iloc[0], "| P(substrate) =", round(float(p_wt), 3))
objectives = [("substrate", "max", "delta_pred"), ("parsimony", "min", "n_mut")]
wild-type: Q14802 | P(substrate) = 0.11
3. Run the optimizer
mode="importance" guides mutations by the static feature importance
(fast, deterministic); run returns the Pareto front df_pareto
(objective columns + non-dominated rank + crowding).
seqo = aa.SeqOpt(mode="importance", model=model, target_class=1, random_state=42)
df_pareto = seqo.run(df_seq=wt, df_feat=df_feat, objectives=objectives,
pop_size=40, n_gen=25, n_mut_max=6, region="tmd")
aa.display_df(df_pareto, n_rows=10, show_shape=True)
DataFrame shape: (7, 8)
| entry | variant | n_mut | sequence_mut | substrate | parsimony | rank | crowding | |
|---|---|---|---|---|---|---|---|---|
| 1 | Q14802 | 0 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 0.000000 | 0.000000 | 0 | inf | |
| 2 | Q14802 | C49V+I54C+I55R+S58R | 4 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 28.000000 | 4.000000 | 0 | inf |
| 3 | Q14802 | S58R | 1 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 14.000000 | 1.000000 | 0 | 0.696429 |
| 4 | Q14802 | C49M+S58R | 2 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 25.000000 | 2.000000 | 0 | 0.482143 |
| 5 | Q14802 | C49V+I55R+S58R | 3 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 27.000000 | 3.000000 | 0 | 0.160714 |
| 6 | Q14802 | Q38W+C49M+S58R | 3 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 27.000000 | 3.000000 | 0 | 0.142857 |
| 7 | Q14802 | C49L+I55R+S58R | 3 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 27.000000 | 3.000000 | 0 | 0.000000 |
df_eval = seqo.eval(df_pareto=df_pareto)
aa.display_df(df_eval, n_rows=10, show_shape=True)
DataFrame shape: (1, 3)
| hypervolume | n_front | spread | |
|---|---|---|---|
| 1 | 66.000000 | 7 | 0.577352 |
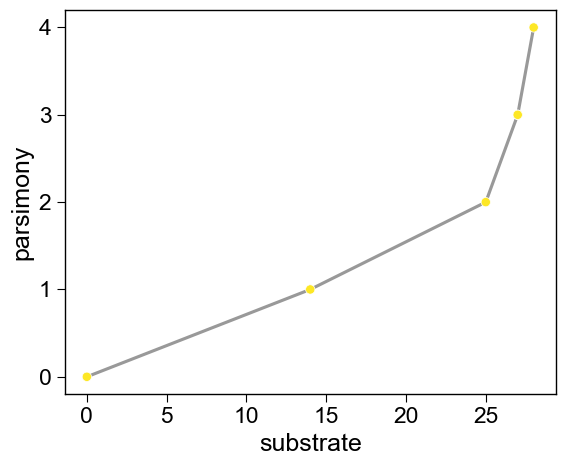
4. Read the trade-off front
Each rank=0 variant is a non-dominated trade-off between substrate
gain and mutation count.
aa.SeqOptPlot().pareto_front(df_pareto=df_pareto, x="substrate", y="parsimony")
plt.tight_layout(); plt.show()
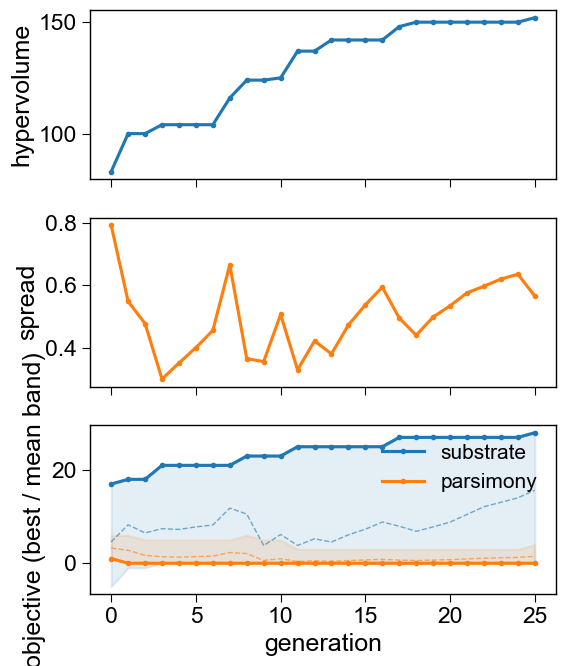
5. Did it converge?
The convergence panels show the dominated hypervolume rising, the front spread, and each objective’s best/mean band across generations.
aa.SeqOptPlot().convergence(history=seqo.history_)
plt.tight_layout(); plt.show()
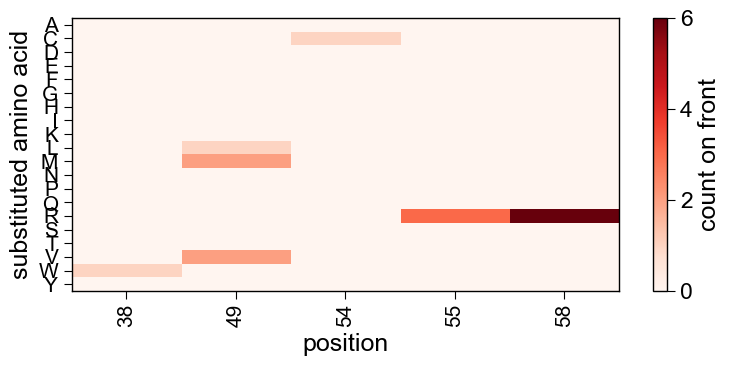
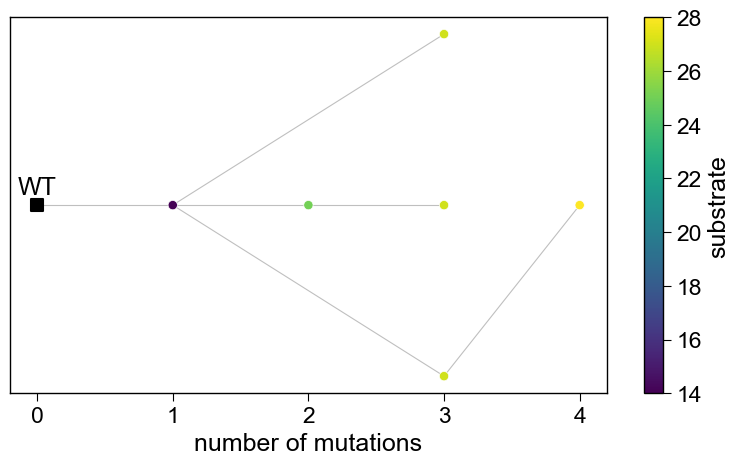
6. Which mutations won, and how were they built up?
The mutation map shows substitution enrichment across the front (position x amino acid); the genealogy shows the mutational lineage from wild-type to the designed variants.
aa.SeqOptPlot().mutation_map(df_pareto=df_pareto)
plt.tight_layout(); plt.show()
aa.SeqOptPlot().genealogy(df_pareto=df_pareto, front_only=False)
plt.tight_layout(); plt.show()
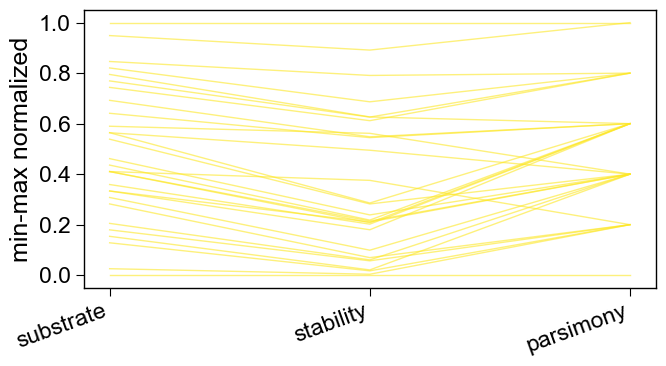
7. More objectives
Add a third objective — keep the feature profile close to natural
(delta_cpp) — and read it with a 3-D front and parallel coordinates.
Objectives can also be any callable(sequence) -> float (a
scikit/torch model, or a sequence-level tool / web API).
objectives3 = [("substrate", "max", "delta_pred"),
("stability", "min", "delta_cpp"),
("parsimony", "min", "n_mut")]
df3 = seqo.run(df_seq=wt, df_feat=df_feat, objectives=objectives3,
pop_size=40, n_gen=25, n_mut_max=6, region="tmd")
aa.SeqOptPlot().parallel_coordinates(df_pareto=df3,
objectives=["substrate", "stability", "parsimony"])
plt.tight_layout(); plt.show()
8. SHAP-guided design
The headline mode="impact" refits a ShapModel every generation
(fuzzy labeling) to mutate the residues SHAP deems most important — pass
the labeled reference set.
df_shap = aa.SeqOpt(mode="impact", model=model, target_class=1, df_seq_ref=df_seq,
labels=labels, random_state=0).run(
df_seq=wt, df_feat=df_feat, objectives=objectives, pop_size=12, n_gen=4,
n_mut_max=4, region="tmd")
aa.display_df(df_shap, n_rows=10, show_shape=True)
DataFrame shape: (9, 8)
| entry | variant | n_mut | sequence_mut | substrate | parsimony | rank | crowding | |
|---|---|---|---|---|---|---|---|---|
| 1 | Q14802 | 0 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 0.000000 | 0.000000 | 0 | inf | |
| 2 | Q14802 | C49E+S58R | 2 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 21.000000 | 2.000000 | 0 | inf |
| 3 | Q14802 | S58F | 1 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 8.000000 | 1.000000 | 0 | 0.559524 |
| 4 | Q14802 | V56M | 1 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 8.000000 | 1.000000 | 0 | 0.440476 |
| 5 | Q14802 | C49E | 1 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 8.000000 | 1.000000 | 0 | 0.000000 |
| 6 | Q14802 | C49E+S58R+A59W | 3 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 20.000000 | 3.000000 | 1 | inf |
| 7 | Q14802 | A50G | 1 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 0.000000 | 1.000000 | 1 | inf |
| 8 | Q14802 | A50G+S58R | 2 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 14.000000 | 2.000000 | 1 | 1.000000 |
| 9 | Q14802 | C49Y+A50G+S58R+A59W | 4 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 20.000000 | 4.000000 | 2 | inf |
Summary
SeqOpt turned a non-substrate into predicted GSEC substrates along a
Pareto front of substrate-gain vs. mutation-count, and SeqOptPlot
exposed the trade-offs, convergence, the enriched mutations and their
lineage. The evolutionary machinery is a pure-Python re-implementation
of NSGA-II (DEAP-equivalent); the design signal comes from your model —
any predict_proba classifier or callable(sequence) predictor.