SeqMutPlot.mutation_landscape
- SeqMutPlot.mutation_landscape(df_scan, entry=None, ax=None, figsize=(12, 6), cmap=None, class_names=None)[source]
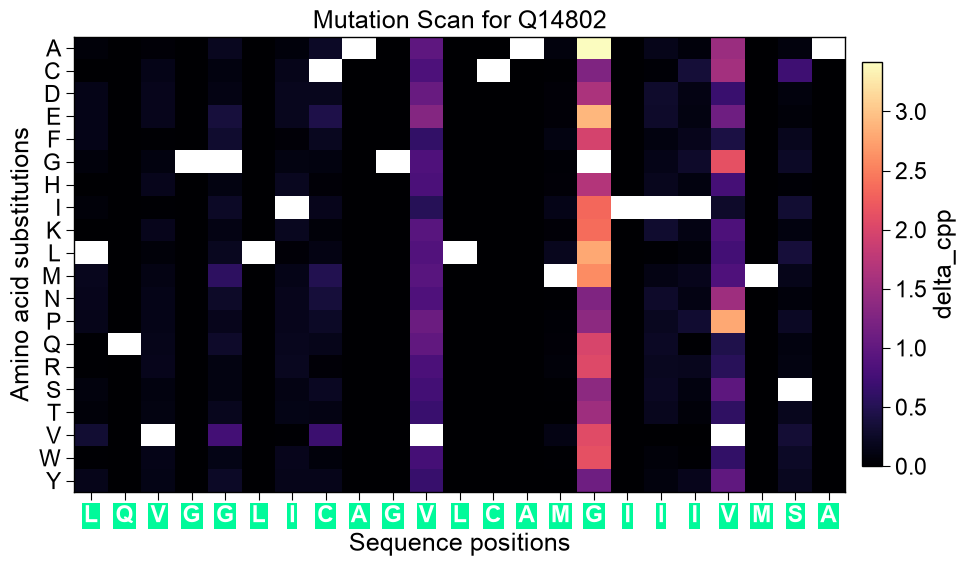
Plot the per-position mutation-scan heatmap for one sequence.
Rows are the 20 canonical amino acids, columns are the scanned positions (labelled by the wild-type residue and colored by sequence part — JMD-N / TMD / JMD-C). Each cell is the change the substitution induces: the model prediction shift
delta_pred(in percentage points, diverging blue-white-red) whenSeqMut.scan()was run with a bound model, otherwise the model-freedelta_cpp. With a model, the title reports the wild-type prediction score.- Parameters:
df_scan (pd.DataFrame) – Mutational landscape produced by
SeqMut.scan().entry (str, optional) – Protein entry to plot. If
None, the first entry indf_scanis used.ax (Axes, optional) – Pre-defined Axes object to plot on. If
None, a new one is created.figsize (tuple, default=(12, 6)) – Figure dimensions (width, height) in inches (used when
axisNone).cmap (str, optional) – Matplotlib colormap name. If
None, a diverging'bwr'is used for the signeddelta_predand'viridis'for the non-negativedelta_cpp.class_names (tuple of str, optional) –
(negative, positive)class labels. When given (and a model was bound), the predicted wild-type class is appended to the title.
- Returns:
fig (Figure) – Figure object containing the plot.
ax (Axes) – Axes object of the mutation-scan heatmap.
Notes
Returned as a
(fig, ax)pair (seeSeqMutPlotfor the shared return contract).
Examples
:meth:
SeqMutPlot.mutation_landscapeis the mutation-scan heatmap: the 20 amino-acid substitutions (rows) against the scanned positions (columns, labelled by the wild-type residue and colored by sequence part). When :meth:SeqMut.scanran with a boundmodel, each cell is the model prediction shiftdelta_pred(diverging blue-white-red) and the title reports the wild-type prediction; otherwise the cells show the model-freedelta_cpp.import itertools import pandas as pd import matplotlib.pyplot as plt import aaanalysis as aa aa.options["verbose"] = False # Data, CPP features, and a fitted TreeModel that scores each sequence df_seq = aa.load_dataset(name="DOM_GSEC", n=10) labels = df_seq["label"].to_list() sf = aa.SequenceFeature() df_parts = sf.get_df_parts(df_seq=df_seq) split_kws = sf.get_split_kws() df_scales = aa.load_scales() cpp = aa.CPP(df_parts=df_parts, split_kws=split_kws, df_scales=df_scales, verbose=False) df_feat = cpp.run(labels=labels, n_filter=25) X = sf.feature_matrix(features=list(df_feat["feature"]), df_parts=df_parts, df_scales=df_scales) tm = aa.TreeModel().fit(X, labels=labels) entry = df_seq["entry"].iloc[0] ts = int(df_seq.set_index("entry").loc[entry, "tmd_start"]) # Model-guided: per-position prediction shift (the mutation-scan heatmap) df_scan = aa.SeqMut(model=tm).scan(df_seq=df_seq, df_feat=df_feat, region=None) aa.plot_settings() aa.SeqMutPlot().mutation_landscape(df_scan=df_scan, entry=entry, class_names=("NEG", "POS"), figsize=(12, 6)) plt.tight_layout() plt.show()

Without a bound model the same method shows the model-free
delta_cppmagnitude.df_scan_free = aa.SeqMut().scan(df_seq=df_seq, df_feat=df_feat, region="tmd") aa.plot_settings() aa.SeqMutPlot().mutation_landscape(df_scan=df_scan_free, entry=entry, figsize=(10, 6)) plt.tight_layout() plt.show()
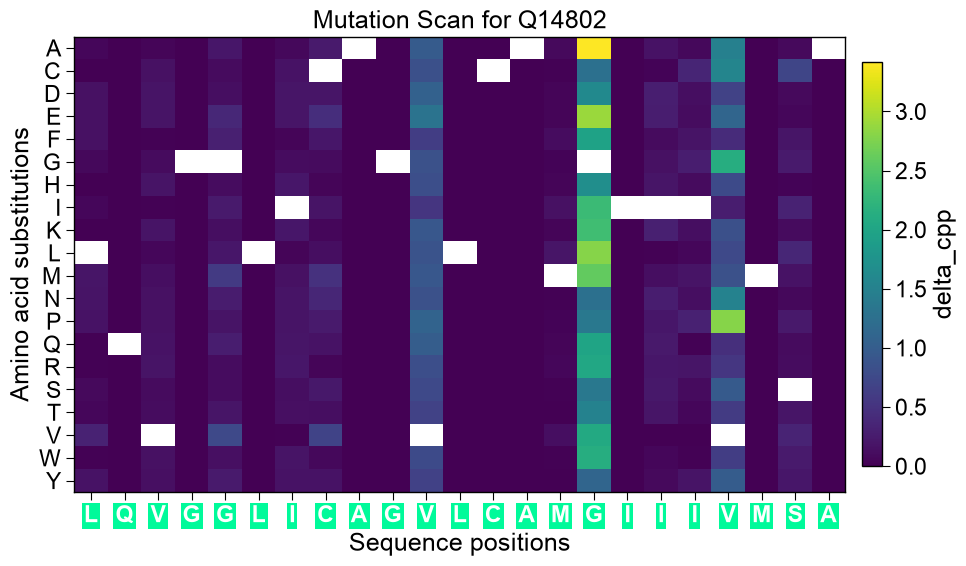
# cmap overrides the default colormap ('bwr' for delta_pred, 'viridis' for delta_cpp) aa.plot_settings() aa.SeqMutPlot().mutation_landscape(df_scan=df_scan_free, entry=entry, figsize=(10, 6), cmap="magma") plt.tight_layout() plt.show()