CPP: Identification of physicochemical signatures
Comparative Physicochemical Profiling (CPP) is a sequence-based algorithm for interpretable feature engineering. It is the centerpiece of AAanalysis, introduced in [Breimann25].
You will learn
Tool:
CPPInput:
df_parts,labels(+split_kwsand scales)Output:
df_feat(the ranked, non-redundant physicochemical signature)Best used for: identifying the physicochemical signature that separates two sequence groups
Related protocol: P1: CPP signature
The aim of the CPP algorithm is to identify a set of unique, non-redundant features that are most discriminant between the test and reference group of sequences. We call this feature set and its visual representation the physicochemical signature of the test group, which can be interpreted at group and sample level with single-residue resolution.
We will demonstrate this in three steps:
Feature Creation
Group Level CPP Analysis
Sample Level CPP Analysis
Feature Creation
To create an CPP object, you just need to provide a valid
df_parts DataFrame:
import aaanalysis as aa
aa.options["verbose"] = False
aa.options["random_state"] = 42
# Load example dataset
df_seq = aa.load_dataset(name="DOM_GSEC")
labels = df_seq["label"].to_list()
sf = aa.SequenceFeature()
# Create Parts
df_parts = sf.get_df_parts(df_seq=df_seq)
# Create CPP object and run with default splits and scales
cpp = aa.CPP(df_parts=df_parts)
A non-redundant set of physicochemical features is obtained through the
CPP.run() method:
# Run CPP algorithm to obtain 100 features (default)
df_feat = cpp.run(labels=labels, n_filter=100)
aa.display_df(df=df_feat, n_rows=8, show_shape=True)
/Users/stephanbreimann/Programming/1Packages/aaanalysis/.claude/worktrees/pr222-fresh/aaanalysis/feature_engineering/_backend/cpp_run.py:143: UserWarning: CPP is using the Python kernel fallback — the compiled Cython extension is not available in this install. Output is bit-exact with the Cython path but ~2x slower. Reinstall via pip install --force-reinstall aaanalysis to fetch a prebuilt wheel. warnings.warn(
DataFrame shape: (100, 13)
| feature | category | subcategory | scale_name | scale_description | abs_auc | abs_mean_dif | mean_dif | std_test | std_ref | p_val_mann_whitney | p_val_fdr_bh | positions | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...2,3)-QIAN880106 | Conformation | α-helix | α-helix (middle) | Weights for alp...ejnowski, 1988) | 0.387000 | 0.118000 | 0.118000 | 0.068000 | 0.080000 | 0.000000 | 0.000000 | 27,28,29,30,31,32,33 |
| 2 | TMD_C_JMD_C-Pat...,14)-CRAJ730103 | Conformation | β-turn | β-turn | Normalized freq...d et al., 1973) | 0.377000 | 0.285000 | -0.285000 | 0.164000 | 0.177000 | 0.000000 | 0.000000 | 27,31 |
| 3 | TMD_C_JMD_C-Seg...6,9)-FAUJ880104 | Shape | Side chain length | Steric parameter | STERIMOL length...e et al., 1988) | 0.367000 | 0.263000 | 0.263000 | 0.161000 | 0.168000 | 0.000000 | 0.000000 | 32,33 |
| 4 | TMD_C_JMD_C-Seg...6,9)-ONEK900101 | Others | Unclassified (Others) | ΔG values in peptides | Delta G values ...-DeGrado, 1990) | 0.366000 | 0.111000 | 0.111000 | 0.070000 | 0.114000 | 0.000000 | 0.000000 | 32,33 |
| 5 | TMD_C_JMD_C-Pat...,15)-QIAN880107 | Conformation | α-helix | α-helix (middle) | Weights for alp...ejnowski, 1988) | 0.363000 | 0.162000 | 0.162000 | 0.091000 | 0.118000 | 0.000000 | 0.000000 | 24,28,32,35 |
| 6 | TMD_C_JMD_C-Seg...3,4)-HUTJ700103 | Energy | Entropy | Entropy | Entropy of form...Hutchens, 1970) | 0.360000 | 0.187000 | 0.187000 | 0.115000 | 0.128000 | 0.000000 | 0.000000 | 31,32,33,34,35 |
| 7 | TMD_C_JMD_C-Seg...2,3)-WOLS870103 | Others | PC 4 | Principal Component 3 (Wold) | Principal prope...d et al., 1987) | 0.359000 | 0.159000 | -0.159000 | 0.090000 | 0.130000 | 0.000000 | 0.000000 | 27,28,29,30,31,32,33 |
| 8 | TMD_C_JMD_C-Pat...,12)-CRAJ730103 | Conformation | β-turn | β-turn | Normalized freq...d et al., 1973) | 0.352000 | 0.227000 | -0.227000 | 0.150000 | 0.170000 | 0.000000 | 0.000000 | 24,28,32 |
CPP Analysis (Group Level)
The CPPPlot class provides various methods for visualizing single to
all features. For further analysis, we need to add the group level
feature importance using the TreeModel class:
# Create feature matrix
sf = aa.SequenceFeature()
X = sf.feature_matrix(features=df_feat["feature"], df_parts=df_parts)
# Fit TreeModel
tm = aa.TreeModel()
tm = tm.fit(X, labels=labels)
# Add feature importance, ranked most-important-first via sort=True
df_feat = tm.add_feat_importance(df_feat=df_feat, sort=True)
The top 15 features can be visualized using the CPPPlot.ranking()
method
import matplotlib.pyplot as plt
# CPP ranking
cpp_plot = aa.CPPPlot()
aa.plot_settings(weight_bold=False, short_ticks=True)
cpp_plot.ranking(df_feat=df_feat)
plt.tight_layout()
plt.show()
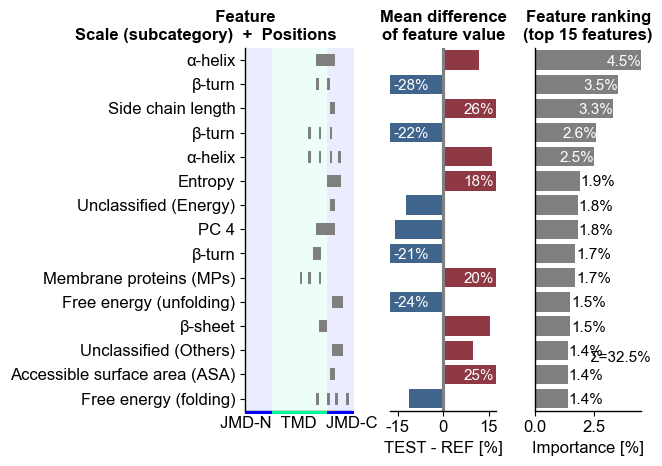
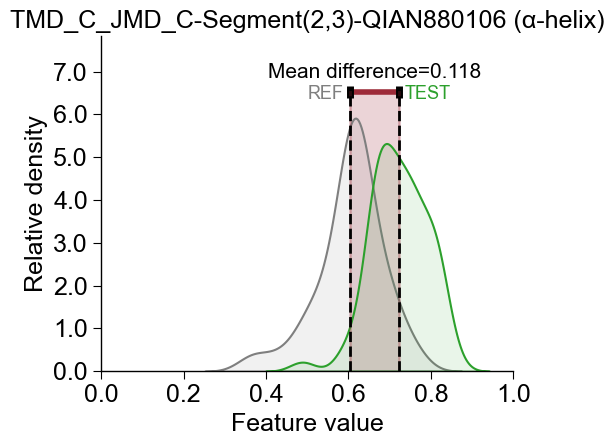
The difference of feature values between the test and the reference
group can be displayed for any selected feature using the
CPPPlot.feature() method:
# df_feat is already sorted by feat_importance (sort=True above), so feat_rank=1 is the top feature
# Show feature value distribution for the best feature
aa.plot_settings()
cpp_plot.feature(feature=df_feat, feat_rank=1, df_seq=df_seq, labels=labels)
plt.title(f"{df_feat['feature'][0]} ({df_feat['subcategory'][0]})")
plt.tight_layout()
plt.show()
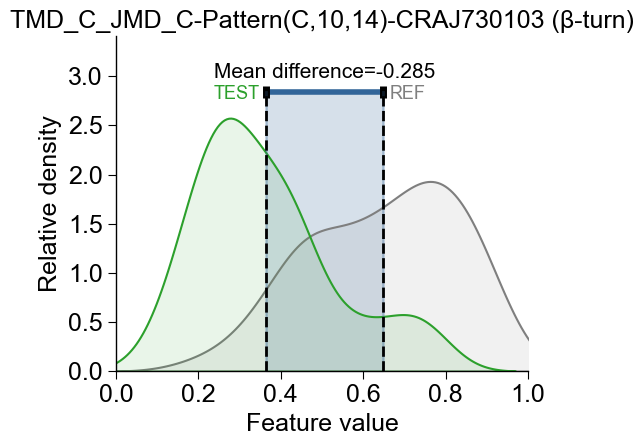
The feature value difference can be either positive (the test dataset has higher values, indicated in red) or negative (the test dataset has lower values, indicated in blue):
# feat_rank=2 selects the second-best feature from the ranked df_feat
aa.plot_settings()
cpp_plot.feature(feature=df_feat, feat_rank=2, df_seq=df_seq, labels=labels)
plt.title(f"{df_feat['feature'][1]} ({df_feat['subcategory'][1]})")
plt.tight_layout()
plt.show()
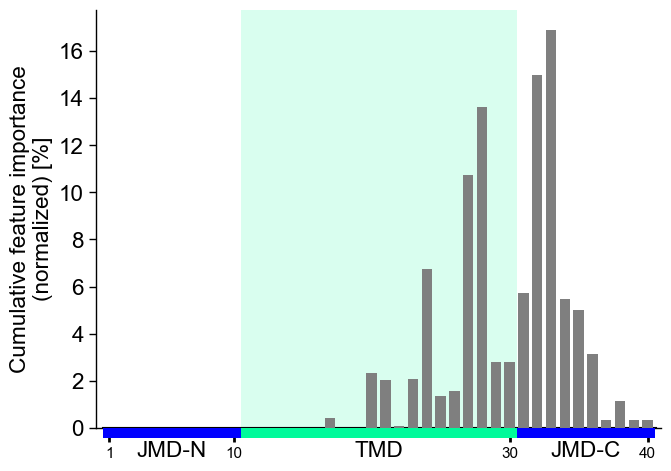
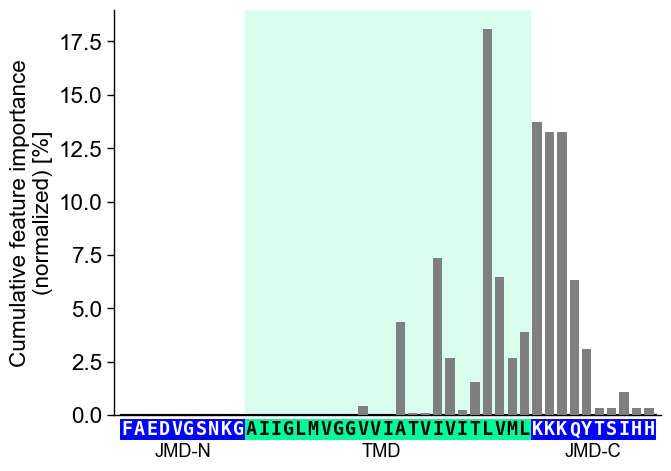
To visualize the importance of all features at single-residue
resolution, the cumulative feature importance per residue position can
be shown using the CPPPlot.profile() method:
# Plot CPP profile
aa.plot_settings(font_scale=0.9)
cpp_plot.profile(df_feat=df_feat)
plt.tight_layout()
plt.show()
The complete feature landscape can be charted using the
CPPPlot.feature_map() method. This CPP feature map shows the feature
value difference and feature importance per residue and scale
subcategory, which are described and discussed in our `AAontology``
(AAontology Usage
Principles,
[Breimann24b]):
# Plot CPP feature map
cpp_plot = aa.CPPPlot()
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.feature_map(df_feat=df_feat)
plt.show()
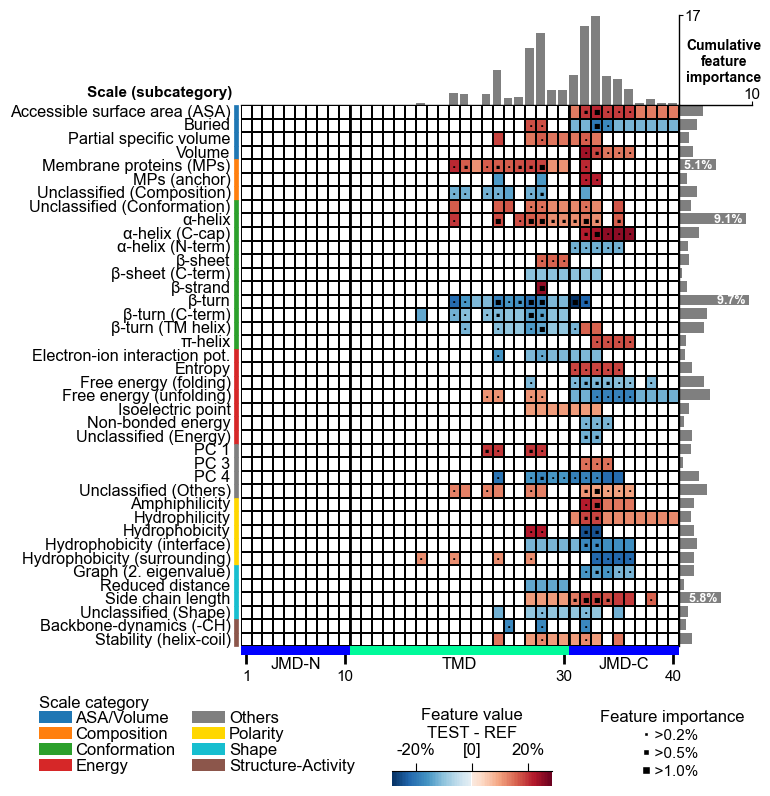
CPP Analysis (“Sample Level”)
You can provide individual sequence parts to the plotting methods to translate the results of the group level CPP features onto a specific sample.
# Get sequences parts for APP
seq_kws = sf.get_seq_kws(df_seq=df_seq, df_parts=df_parts, sample="P05067")
Provide the parts as tmd_seq, jmd_n_seq, and jmd_c_seq
parameters to the CPPPlot.profile() method:
# Plot CPP profile ("sample level")
aa.plot_settings(font_scale=0.9)
cpp_plot.profile(df_feat=df_feat, **seq_kws)
plt.tight_layout()
plt.show()
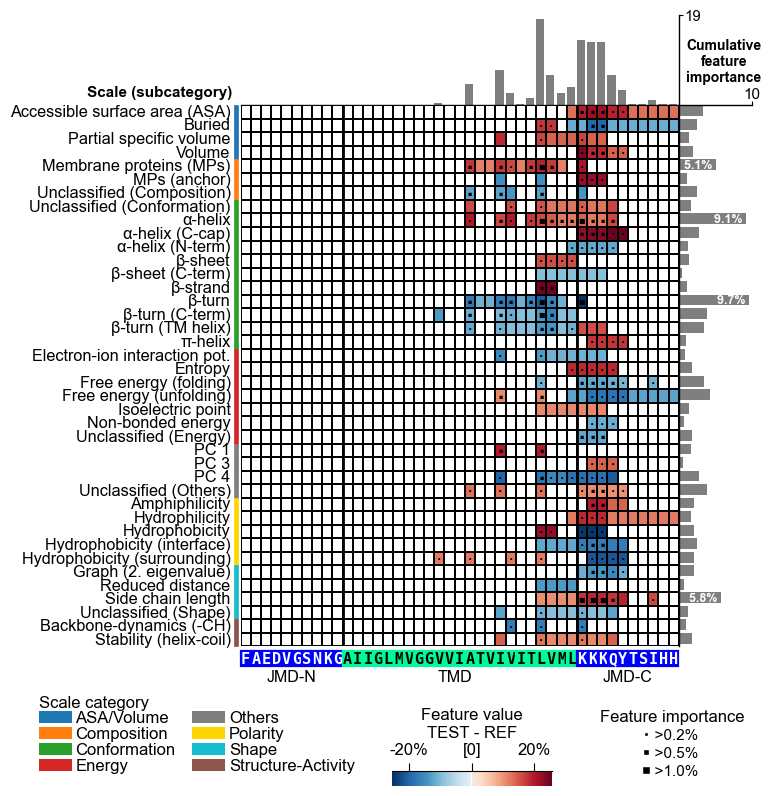
Or to the feature_map() method:
# Plot CPP feature map ("sample level")
cpp_plot = aa.CPPPlot()
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.feature_map(df_feat=df_feat, **seq_kws)
plt.show()
However, these are still the general results but only visualized for a specific sample sequence. To obtain the sample-specific feature value difference and feature impact, see the Explainable AI Tutorial.
For more details on the CPP and CPPPlot classes are given in the
Feature Engineering
API.
To delve deeper into the feature concept, see the CPP Usage
Principles
section.