ShapModel: Explaining with single-residue resolution
To enable explanations of sample-specific predictions at the
single-residue level, we’ve developed a wrapper model around SHAP
(SHapley Additive
exPlanations).
This explainable AI framework adopts a game-theoretic approach to
elucidate the output of any machine learning model. Our ShapModel
model, introduced in [Breimann25], fits multiple SHAP explainers and
integrates their output to provide a robust estimate of feature impacts.
You will learn
Tool:
ShapModelInput: a feature matrix
X,labels, and one or more model classesOutput: per-sample SHAP feature impact (a
df_featimpact column)Best used for: explaining individual predictions at single-residue resolution
Related protocol: P9: Interpretability
Related API:
ShapModel
Obtaining Feature Impact
To illustrate, we’ll use an example dataset comprising γ-secretase substrates (n=63) and non-substrates (n=63) from [Breimann25]. We’ll explain the model’s prediction output for the Alzheimer’s disease-associated amyloid precursor protein (APP):
import aaanalysis as aa
aa.options["verbose"] = False
aa.options["random_state"] = 42
# Load dataset and respective features
df_seq = aa.load_dataset(name="DOM_GSEC")
labels = list(df_seq["label"])
df_feat = aa.load_features(name="DOM_GSEC")
# Show APP
aa.display_df(df=df_seq, n_rows=10, char_limit=25)
| entry | gene | sequence | label | tmd_start | tmd_stop | jmd_n | tmd | jmd_c | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | P05067 | APP | MLPGLALLLLAA...NPTYKFFEQMQN | 1 | 701 | 723 | FAEDVGSNKG | AIIGLMVGGVVIATVIVITLVML | KKKQYTSIHH |
| 2 | P14925 | Pam | MAGRARSGLLLL...YSAPLPKPAPSS | 1 | 868 | 890 | KLSTEPGSGV | SVVLITTLLVIPVLVLLAIVMFI | RWKKSRAFGD |
| 3 | P70180 | Npr3 | MRSLLLFTFSAC...REDSIRSHFSVA | 1 | 477 | 499 | PCKSSGGLEE | SAVTGIVVGALLGAGLLMAFYFF | RKKYRITIER |
| 4 | Q03157 | Aplp1 | MGPTSPAARGQG...ENPTYRFLEERP | 1 | 585 | 607 | APSGTGVSRE | ALSGLLIMGAGGGSLIVLSLLLL | RKKKPYGTIS |
| 5 | Q06481 | APLP2 | MAATGTAAAAAT...NPTYKYLEQMQI | 1 | 694 | 716 | LREDFSLSSS | ALIGLLVIAVAIATVIVISLVML | RKRQYGTISH |
| 6 | P35613 | BSG | MAAALFVLLGFA...DKGKNVRQRNSS | 1 | 323 | 345 | IITLRVRSHL | AALWPFLGIVAEVLVLVTIIFIY | EKRRKPEDVL |
| 7 | P35070 | BTC | MDRAARCSGASS...PINEDIEETNIA | 1 | 119 | 141 | LFYLRGDRGQ | ILVICLIAVMVVFIILVIGVCTC | CHPLRKRRKR |
| 8 | P09803 | Cdh1 | MGARCRSFSALL...KLADMYGGGEDD | 1 | 711 | 733 | GIVAAGLQVP | AILGILGGILALLILILLLLLFL | RRRTVVKEPL |
| 9 | P19022 | CDH2 | MCRIAGALRTLL...KKLADMYGGGDD | 1 | 724 | 746 | RIVGAGLGTG | AIIAILLCIIILLILVLMFVVWM | KRRDKERQAK |
| 10 | P16070 | CD44 | MDKFWWHAAWGL...RNLQNVDMKIGV | 1 | 650 | 672 | GPIRTPQIPE | WLIILASLLALALILAVCIAVNS | RRRCGQKKKL |
To fit the ShapModel, we fist need to create the feature matrix
using the SequenceFeature.feat_matrix() method:
# Create feature matrix
sf = aa.SequenceFeature()
df_parts = sf.get_df_parts(df_seq=df_seq)
X = sf.feature_matrix(df_parts=df_parts, features=df_feat["feature"])
The difference of feature values between a selected protein (e.g., APP)
and the reference dataset can be included into the feature DataFrame
using the ShapModel.add_sample_mean_dif() method. This will create a
new mean_dif_'name' column (e.g., ‘mean_dif_APP’):
sm = aa.ShapModel()
sm.fit(X, labels=labels)
# Add the feature value difference for the first protein (APP)
df_feat = sm.add_sample_mean_dif(X, labels=labels, df_feat=df_feat,
samples=0, names="APP")
To add the sample-specific feature impact, use the
ShapModel.add_feat_impact() method. This will create a new
feat_impact_'name' column (e.g., ‘feat_impact_APP’):
# Add feature impacts for the first protein (APP)
df_feat = sm.add_feat_impact(df_feat=df_feat, samples=0, names="APP")
aa.display_df(df=df_feat, n_rows=5)
| feature | category | subcategory | scale_name | scale_description | abs_auc | abs_mean_dif | mean_dif | std_test | std_ref | p_val_mann_whitney | p_val_fdr_bh | positions | feat_importance | feat_importance_std | mean_dif_APP | feat_impact_APP | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...3,4)-KLEP840101 | Energy | Charge | Charge | Net charge (Kle...n et al., 1984) | 0.244000 | 0.103666 | 0.103666 | 0.106692 | 0.110506 | 0.000000 | 0.000000 | 31,32,33,34,35 | 0.970400 | 1.438918 | 0.220635 | 0.980000 |
| 2 | TMD_C_JMD_C-Seg...3,4)-FINA910104 | Conformation | α-helix (C-cap) | α-helix termination | Helix terminati...n et al., 1991) | 0.243000 | 0.085064 | 0.085064 | 0.098774 | 0.096946 | 0.000000 | 0.000000 | 31,32,33,34,35 | 0.000000 | 0.000000 | 0.193990 | 1.320000 |
| 3 | TMD_C_JMD_C-Seg...6,9)-LEVM760105 | Shape | Side chain length | Side chain length | Radius of gyrat... (Levitt, 1976) | 0.233000 | 0.137044 | 0.137044 | 0.161683 | 0.176964 | 0.000000 | 0.000001 | 32,33 | 1.554800 | 2.109848 | 0.283275 | 1.710000 |
| 4 | TMD_C_JMD_C-Seg...3,4)-HUTJ700102 | Energy | Entropy | Entropy | Absolute entrop...Hutchens, 1970) | 0.229000 | 0.098224 | 0.098224 | 0.106865 | 0.124608 | 0.000000 | 0.000001 | 31,32,33,34,35 | 3.111200 | 3.109955 | 0.162838 | 2.870000 |
| 5 | TMD_C_JMD_C-Seg...6,9)-RADA880106 | ASA/Volume | Volume | Accessible surface area (ASA) | Accessible surf...olfenden, 1988) | 0.223000 | 0.095071 | 0.095071 | 0.114758 | 0.132829 | 0.000000 | 0.000002 | 32,33 | 0.000000 | 0.000000 | 0.189680 | 1.140000 |
Visualizing Feature Impact
Features can have either a positive (red) or negative (blue) impact, indicating whether they increase or decrease the model’s prediction output, respectively. Before visualizing impact of features, we need to obtain its sequence parts:
import matplotlib.pyplot as plt
# Get sequences parts for APP
seq_kws = sf.get_seq_kws(df_seq=df_seq, df_parts=df_parts, sample="P05067") # Accession number of APP
To explain the feature impact with single-residue resolution, AAanalysis offers the following four types of visualizations: CPP-SHAP ranking plot, CPP-SHAP profile, CPP heatmap, CPP-SHAP heatmap. An overview is provided under Explainable AI Usage Principles
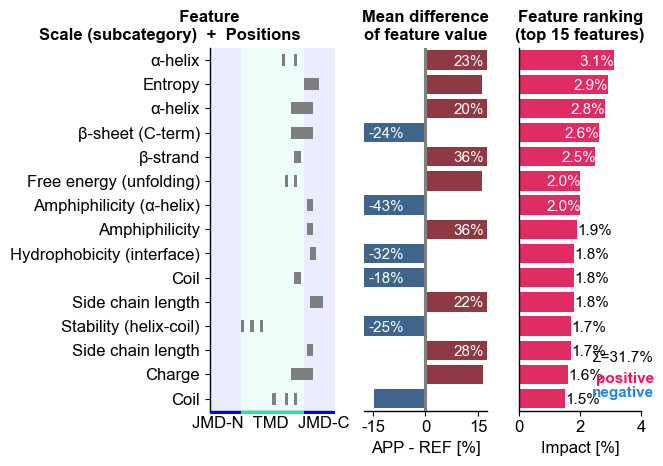
We can first show the feature ranking for a selected protein
(‘Protein0’) using the CPPPlot.ranking() method, plotting SHAP
analysis results by setting shap_plot=True:
# CPP-SHAP ranking plot
cpp_plot = aa.CPPPlot()
aa.plot_settings(short_ticks=True, weight_bold=False)
cpp_plot.ranking(df_feat=df_feat, shap_plot=True,
col_dif="mean_dif_APP", col_imp="feat_impact_APP",
name_test="APP")
plt.tight_layout()
plt.show()
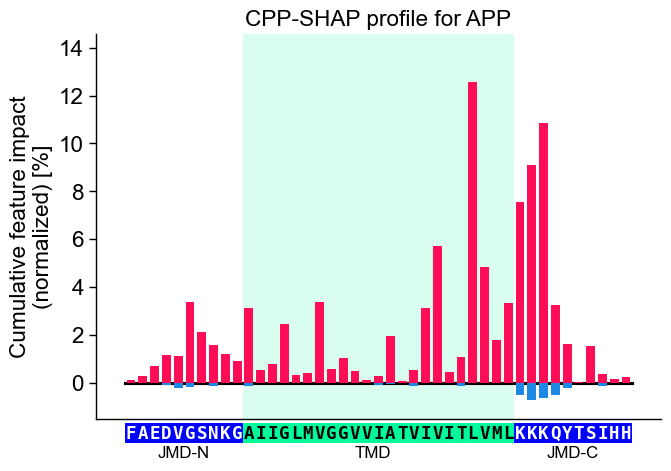
Show the specific CPP-SHAP Profile for the first Protein using the
CPPPlot.profile() method with setting shap_plot=True:
# CPP-SHAP profile
aa.plot_settings(font_scale=0.9)
cpp_plot.profile(df_feat=df_feat, shap_plot=True,
col_imp="feat_impact_APP", **seq_kws)
plt.title("CPP-SHAP profile for APP")
plt.tight_layout()
plt.show()
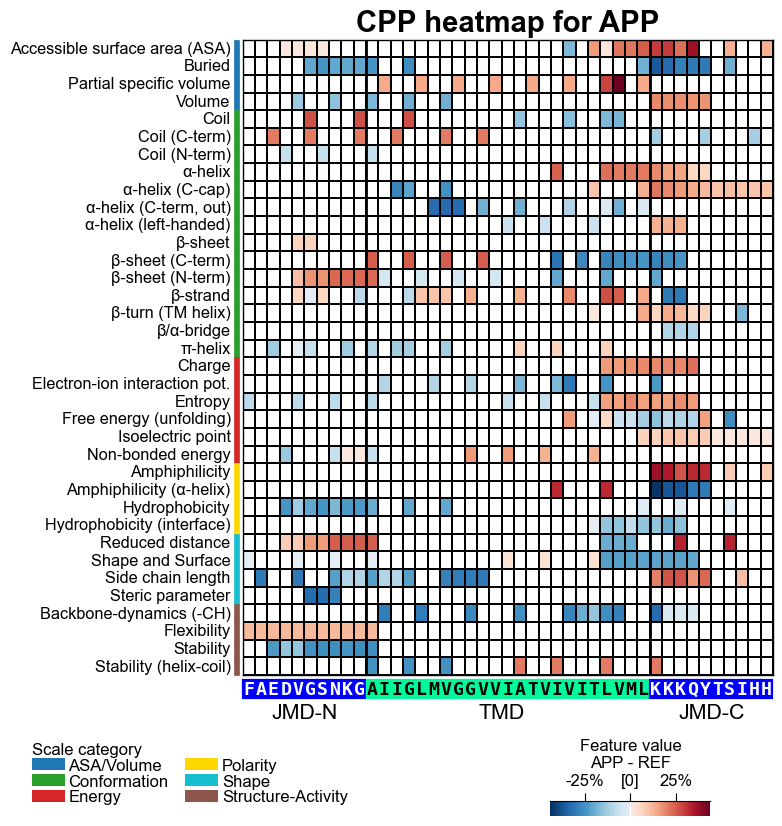
With the CPPPlot.heatmap() method we can visualize the
sample-specific feature value difference and the feature impact per
scale subcategory and residue position. Set shap_plot=True and
provide the respective column with the mean difference and the feature
impact:
# CPP heatmap (sample level)
fs = aa.plot_gcfs()
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.heatmap(df_feat=df_feat, shap_plot=True,
col_val="mean_dif_APP", **seq_kws, name_test="APP")
plt.title("CPP heatmap for APP", fontsize=fs+5, weight="bold")
plt.show()
# CPP-SHAP heatmap (sample level)
cpp_plot.heatmap(df_feat=df_feat, shap_plot=True,
col_val="feat_impact_APP", **seq_kws)
plt.title("CPP-SHAP heatmap for APP", fontsize=fs+5, weight="bold")
plt.show()
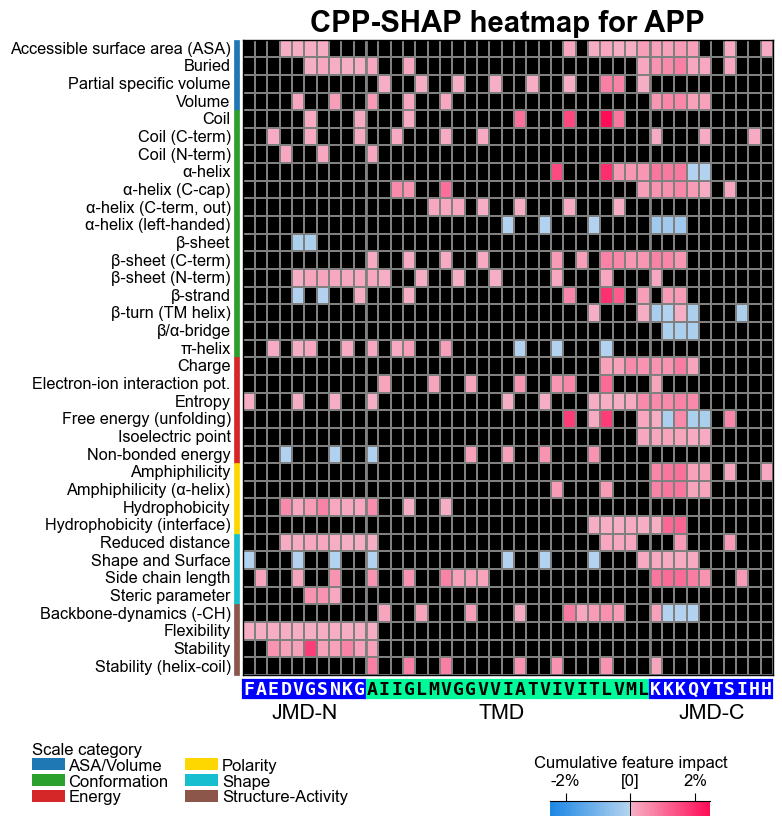
To compare a specific sample, such as APP, with the positive test class, re-compute the feature value differences between the sample and the test set and visualize them as a CPP heatmap. We do not recommend using the feature map here, as feature impact or importance would not properly reflect the machine learning training context. Therefore, another colormap (‘PiYG_r’) is used to indicate the feature value differences.
# Add the feature value difference for the first protein (APP) against the test group
df_feat_app = sm.add_sample_mean_dif(X, labels=labels, df_feat=df_feat,
samples=0, names="APP", label_ref=1, drop=True)
# CPP-SHAP heatmap (sample level vs Test)
cpp_plot.heatmap(df_feat=df_feat_app, shap_plot=True,
col_val="mean_dif_APP", **seq_kws, name_test="APP", name_ref="Test set",
cmap="PiYG_r")
plt.title("CPP heatmap for APP vs Test set", fontsize=fs+5, weight="bold")
plt.show()
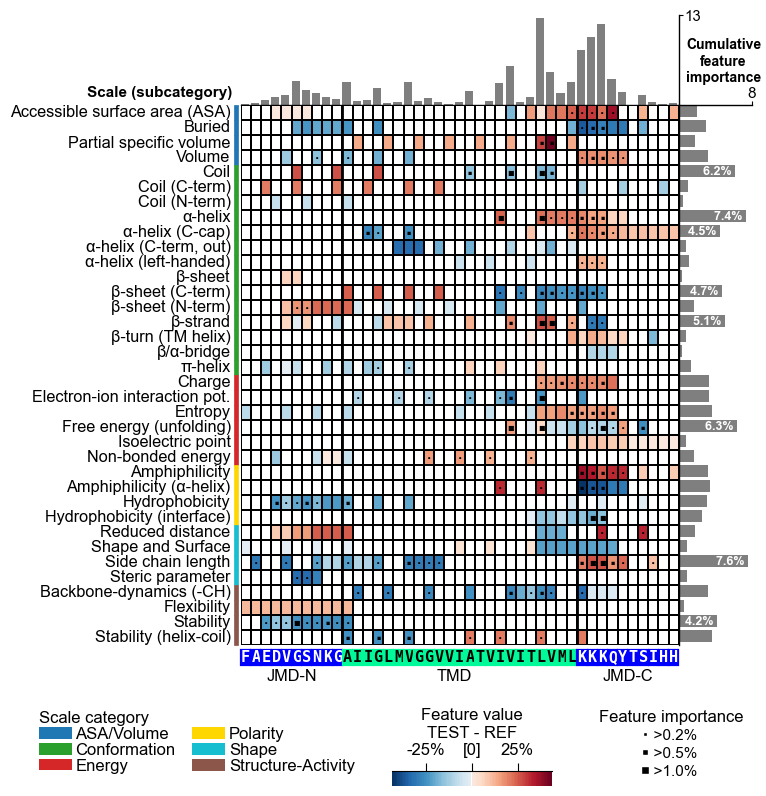
You can create as well the feature map for individual samples. We first have the convert the feature impact into feature importance by simply taking their absolute values.
# Add feature importance
df_feat.insert(len(df_feat.T), "feat_importance_APP", [abs(x) for x in df_feat["feat_impact_APP"]])
The feature map can be created using the CPPPlot.feature_map()
method with providing the respective sequence parts and data columns.
You can also set shap_plot=True and provide the respective column
with the mean difference and the feature impact:
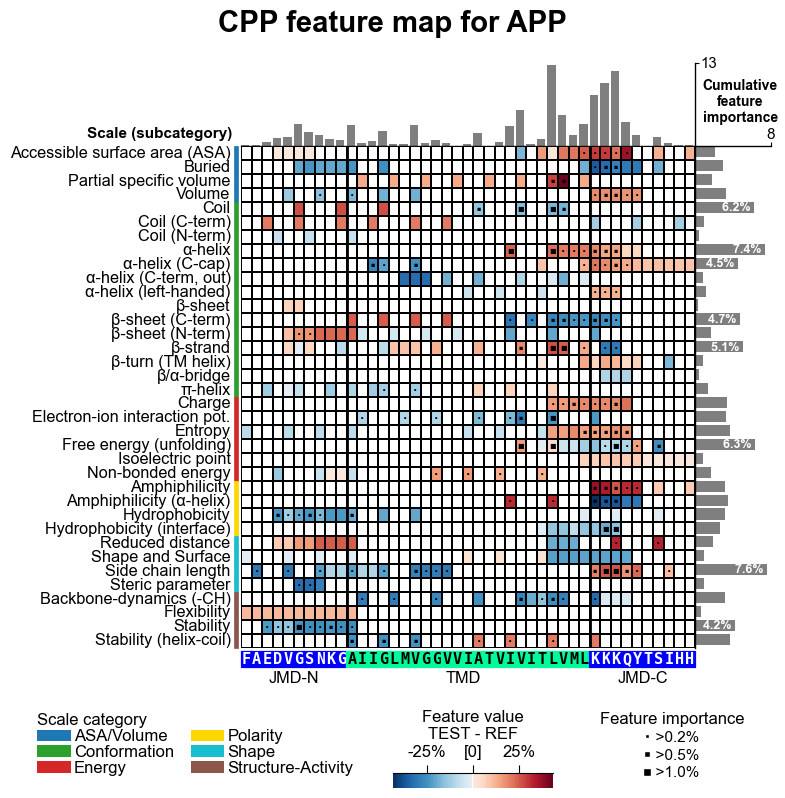
# CPP feature map (sample level with feature importance)
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.feature_map(df_feat=df_feat, col_val="mean_dif_APP", col_imp="feat_importance_APP", **seq_kws)
plt.suptitle("CPP feature map for APP", fontsize=fs+5, weight="bold")
plt.tight_layout()
plt.show()
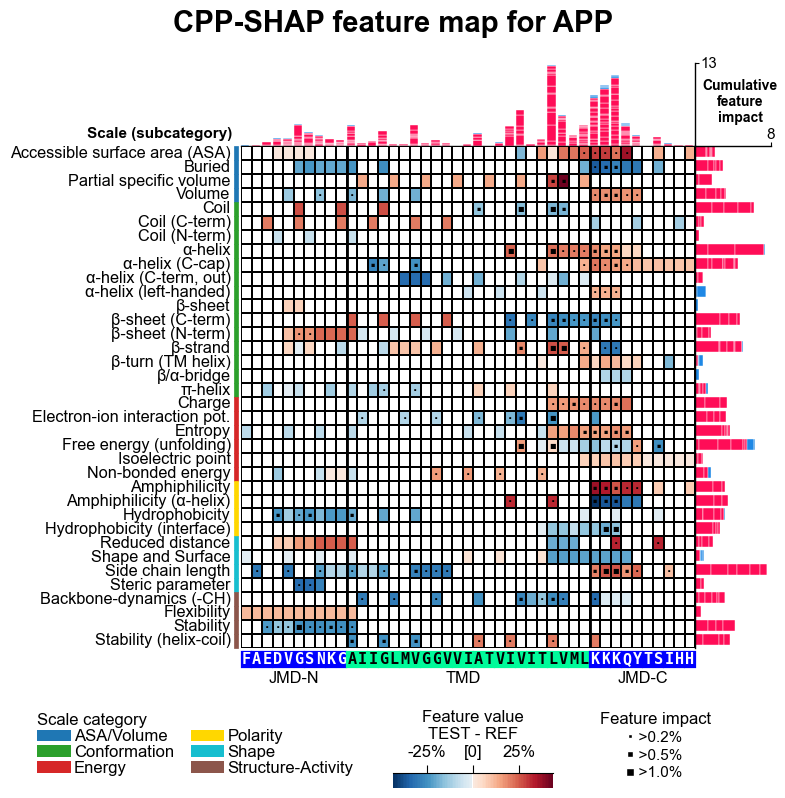
# CPP feature map (sample level with feature impact)
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot.feature_map(df_feat=df_feat, col_val="mean_dif_APP", col_imp="feat_impact_APP", shap_plot=True, **seq_kws)
plt.suptitle("CPP-SHAP feature map for APP", fontsize=fs+5, weight="bold")
plt.tight_layout()
plt.show()
Details on the ShapModel class can be found in the ShapModel
API.
More information on explainable AI and how CPP and SHAP were combined
are provided in the Explainable AI Usage
Principles
section.