P9: Interpretability
Turn a group-level CPP signature into a per-sample, single-residue
explanation: SHAP and the CPP-SHAP heatmap. A CPP signature (P1)
tells you which position-resolved physicochemical features separate a
test group (label=1) from a reference group (label=0)
on average. This protocol goes one level deeper: given that signature
and a fitted tree model, ShapModel computes SHapley Additive
exPlanations (SHAP) values, the signed contribution of each feature to
one protein’s prediction score. The sample-level heatmap()
(with shap_plot=True) then anchors those signed attributions onto
the real residue positions of a single protein, so the explanation reads
like “this protein’s TMD hydrophobicity at this position raised its
substrate score” rather than an abstract feature index.
The signature is the group story; SHAP is the per-protein story. Feature importance is unsigned and group-level (how much does this feature matter overall?); feature impact is signed and sample-level (how much, and in which direction, did this feature push the score for this one protein?): red pushes toward the test class, blue toward the reference class. Read one sample’s impact as a local explanation, not as a new group claim.
We stay at the domain level (dataset prefix DOM_): the unit of
comparison is the transmembrane-domain (TMD) part set of the
gamma-secretase substrate task (DOM_GSEC), and we explain a single
protein, APP, within it.
When to use it. Use this protocol when you already have a CPP
signature (df_feat) plus a labelled feature matrix, and you
want to explain a single protein’s prediction rather than the group as
a whole. The biological question shifts from “what distinguishes
substrates from non-substrates?” (determinant discovery, P1) to:
“Why does the model classify **this* protein (e.g. APP) as a gamma-secretase substrate, and where in its sequence do the decisive physicochemical differences act?”*
This is the protein-prediction analogue of a feature-attribution map (LIME / SHAP) in deep learning, but every attribution is anchored to an interpretable physicochemical scale and to a concrete residue position, not a black-box embedding dimension.
When not to use it. SHAP explains; it does not establish trust.
Don’t use a single sample’s feature impact to make a group claim
(“this scale defines substrates”): that is the signature’s job (P1).
Don’t read it as proof the model generalizes: attributions are sample-
and model-specific, and a model fit on a tiny set can attribute
confidently to noise, so stability and controls come next (P10). And
skip it entirely if you only need a ranked group signature:
add_feat_importance() already gives the unsigned,
group-level ranking without fitting SHAP.
Input. This protocol receives its signature from P1: CPP signature and a fitted tree model from P7: Build an interpretable classifier. Concretely it needs:
``df_seq`` with a binary
labelcolumn (test group = 1, reference group = 0). Here: the bundledDOM_GSECset (gamma-secretase substrates vs. non-substrates), a domain-level task over the TMD and its flanking juxtamembrane (JMD) parts.``df_feat``, the CPP signature from P1 (
aa.load_features(name="DOM_GSEC")provides a precomputed signature here so this protocol stays self-contained).``X``, the
(n_samples, n_features)feature matrix built fromdf_feat["feature"]viafeature_matrix().``labels``, the class vector aligned to the rows of
X.
We pick one protein to explain: APP (UniProt P05067), the
Alzheimer’s-disease amyloid precursor protein and a canonical
gamma-secretase substrate.
import aaanalysis as aa
import matplotlib.pyplot as plt
aa.options["verbose"] = False
aa.options["random_state"] = 42
# Small fixture: 50 per class (100 proteins total) keeps SHAP fitting fast
df_seq = aa.load_dataset(name="DOM_GSEC", n=50)
labels = df_seq["label"].to_list()
# CPP signature (from Protocol 1) and the feature matrix it indexes
df_feat = aa.load_features(name="DOM_GSEC")
sf = aa.SequenceFeature()
df_parts = sf.get_df_parts(df_seq=df_seq)
X = sf.feature_matrix(df_parts=df_parts, features=df_feat["feature"])
# Locate APP's row in X (non-substrates come first, so APP is NOT index 0; it sits at 50)
pos_app = list(df_seq["entry"]).index("P05067")
Run. Four steps, all on the real API (see the ShapModel tutorial for the function details):
Fit :class:`~aaanalysis.ShapModel`: trains the tree models and runs the SHAP explainer over several Monte-Carlo rounds, storing the per-sample attributions in
shap_values.Add the feature value difference for APP (
add_sample_mean_dif): how APP’s feature values differ from the reference-group average.Add the feature impact for APP (
add_feat_impact): the signed, normalized SHAP attribution per feature.Visualize with the sample-level
CPPPlotmethods (shap_plot=True).
# Fit ShapModel (pro feature; shap ships with the `pro` extra)
try:
sm = aa.ShapModel(verbose=False, random_state=42)
sm = sm.fit(X, labels=labels, n_rounds=3) # 3 rounds is enough for a demo fixture
except ImportError as e:
raise ImportError("ShapModel requires shap: pip install aaanalysis[pro]") from e
# Feature value difference: APP vs. the reference (non-substrate) group average
df_feat = sm.add_sample_mean_dif(X, labels=labels, df_feat=df_feat,
samples=pos_app, names="APP")
# Signed SHAP feature impact for APP (normalized to % of total absolute impact)
df_feat = sm.add_feat_impact(df_feat=df_feat, samples=pos_app, names="APP")
aa.display_df(df=df_feat[["feature", "category", "mean_dif_APP", "feat_impact_APP"]], n_rows=8)
| feature | category | mean_dif_APP | feat_impact_APP | |
|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...3,4)-KLEP840101 | Energy | 0.224000 | 0.660000 |
| 2 | TMD_C_JMD_C-Seg...3,4)-FINA910104 | Conformation | 0.194644 | 1.280000 |
| 3 | TMD_C_JMD_C-Seg...6,9)-LEVM760105 | Shape | 0.300050 | 1.570000 |
| 4 | TMD_C_JMD_C-Seg...3,4)-HUTJ700102 | Energy | 0.177140 | 3.050000 |
| 5 | TMD_C_JMD_C-Seg...6,9)-RADA880106 | ASA/Volume | 0.201494 | 1.300000 |
| 6 | TMD_C_JMD_C-Seg...2,3)-KLEP840101 | Energy | 0.167146 | 1.250000 |
| 7 | TMD_C_JMD_C-Seg...4,5)-FAUJ880109 | Energy | 0.146250 | 0.210000 |
| 8 | TMD_C_JMD_C-Seg...3,4)-JANJ780101 | ASA/Volume | 0.373808 | 0.540000 |
# APP's sequence parts, used to anchor the plots to real residues
seq_kws = sf.get_seq_kws(df_seq=df_seq, df_parts=df_parts, sample="P05067")
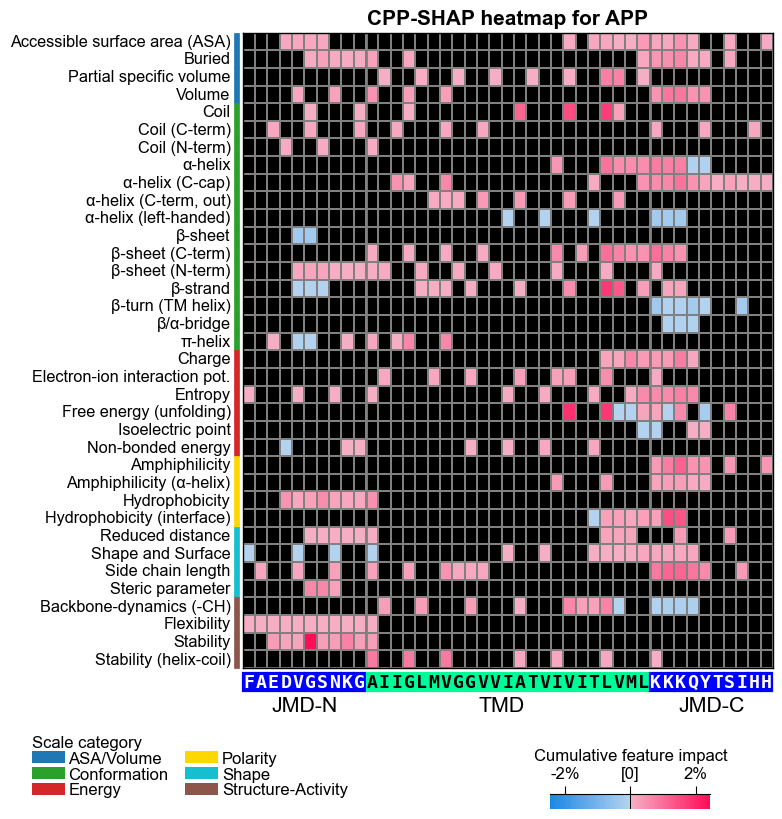
# CPP-SHAP heatmap (sample level): the headline figure.
# Each cell is the SIGNED SHAP impact of one feature on APP's substrate score,
# anchored to its real residue position and scale subcategory
# (red = pushes APP toward "substrate", blue = pushes toward "non-substrate").
fs = aa.plot_gcfs()
aa.plot_settings(font_scale=0.65, weight_bold=False)
cpp_plot = aa.CPPPlot()
fig, ax = cpp_plot.heatmap(df_feat=df_feat, shap_plot=True,
col_val="feat_impact_APP", name_test="APP", **seq_kws)
plt.title("CPP-SHAP heatmap for APP", fontsize=fs + 5, weight="bold")
plt.show()
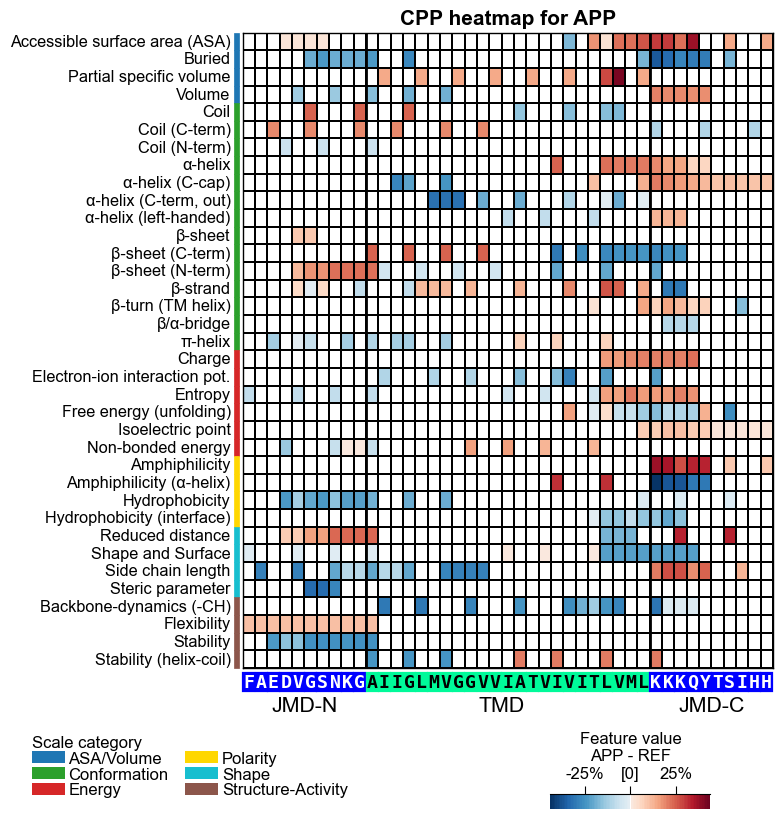
# CPP heatmap (sample level, mean-difference mode):
# each cell is APP's feature VALUE difference vs. the reference-group average,
# per scale subcategory and residue position (the complement to the impact heatmap above).
aa.plot_settings(font_scale=0.65, weight_bold=False)
fig, ax = cpp_plot.heatmap(df_feat=df_feat, shap_plot=True,
col_val="mean_dif_APP", name_test="APP", **seq_kws)
plt.title("CPP heatmap for APP", fontsize=fs + 5, weight="bold")
plt.show()
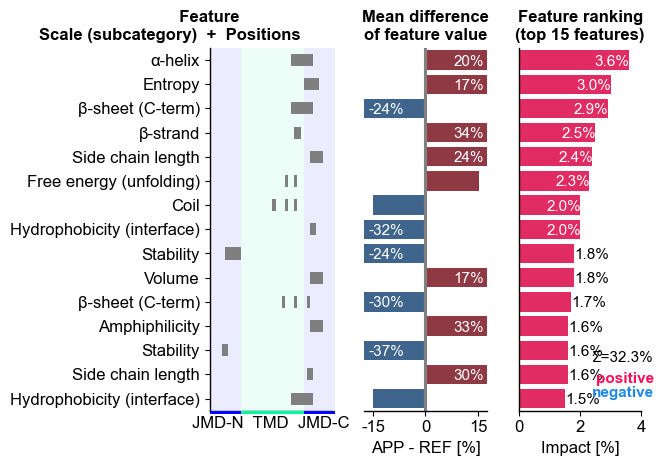
# CPP-SHAP ranking: top features for APP ordered by impact magnitude
aa.plot_settings(short_ticks=True, weight_bold=False)
fig, ax = cpp_plot.ranking(df_feat=df_feat, shap_plot=True,
col_dif="mean_dif_APP", col_imp="feat_impact_APP",
name_test="APP")
plt.tight_layout()
plt.show()
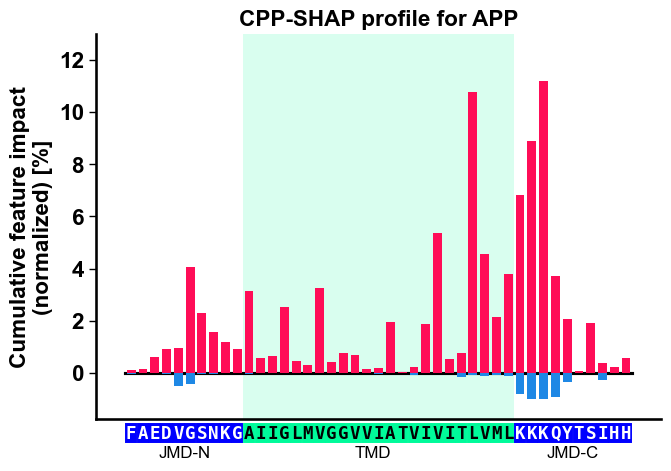
# CPP-SHAP profile: cumulative signed impact per residue position along APP
aa.plot_settings(font_scale=0.9)
fig, ax = cpp_plot.profile(df_feat=df_feat, shap_plot=True,
col_imp="feat_impact_APP", **seq_kws)
plt.title("CPP-SHAP profile for APP")
plt.tight_layout()
plt.show()
Output. df_feat gains two sample-level columns:
mean_dif_APP: APP’s feature value difference vs. the reference-group average (sign = direction).feat_impact_APP: the signed SHAP attribution per feature, normalized so the absolute values sum to 100%.
Sample-level CPP-SHAP figures:
CPP-SHAP heatmap (impact mode): the headline figure; each cell is the signed SHAP impact of one feature on APP’s score (diverging colormap, red positive / blue negative), per scale subcategory and residue position.
CPP heatmap (mean-difference mode): each cell is APP’s feature value difference vs. the reference-group average, the complement that shows where APP physically differs.
Ranking: APP’s top features ordered by impact magnitude.
Profile: cumulative signed impact along the residue positions of APP’s JMD-N / TMD / JMD-C.
Every figure is anchored to APP’s actual sequence (jmd_n, tmd,
jmd_c), so an attribution can be read off a specific residue.
How to interpret. A few things are happening in these figures. The heatmap row tells you which physicochemical subcategory and where along APP’s parts; the color tells you direction; the cumulative bars sum the signed impact per residue position. Read them like this:
Element |
Reading |
|---|---|
red bar / cell |
feature’s SHAP impact is positive, pushing APP toward the test group (substrate) |
blue bar / cell |
feature’s SHAP impact is negative, pushing APP toward the reference group (non-substrate) |
tall / saturated segment |
feature contributes strongly in that direction |
bar width at position x |
cumulative impact from all features acting at residue position x |
heatmap cell, mean-diff mode |
APP’s feature value difference (test minus reference) at one position/scale |
heatmap cell, impact mode |
the signed contribution of that one feature to APP’s score |
Worked reading (a recipe, not a claim about APP). This is the template you apply to whatever your own figure shows. For example, if you see a tall red bar over a TMD position whose row is a hydrophobicity subcategory, read it as: APP’s hydrophobicity is higher than the reference-group average there, and that excess hydrophobicity raised the classifier’s substrate score for APP. Swap in the actual subcategory, position, and color you observe: the figure, not this sentence, is the source of truth.
Key takeaways
Coherent same-color blocks (e.g. red hydrophobicity across the TMD core) are a consistent, reliable local signal; isolated single cells are weak or conflicting evidence, so don’t over-read them.
Sign agreement between
mean_dif_APPandfeat_impact_APPmeans APP follows the group trend the model learned (a confidence boost); a sign mismatch flags a possible outlier or a non-linear effect, so interpret with care.This is one protein’s local explanation. A high-impact feature for APP is not automatically a group determinant, so cross-check against the signature (P1) before generalizing.
Common mistakes.
Calling ``add_feat_impact`` before ``fit``:
shap_valuesis unset untilfit()runs, so impact cannot be computed.Assuming the sample is at index 0: with
load_dataset(name="DOM_GSEC", n=50)the reference class (label 0) comes first, so APP sits at position 50, not 0. Always resolve the position from the entry (herelist(df_seq["entry"]).index("P05067")).Mismatching ``shap_plot`` and the column: when
shap_plot=True,col_valmust be a per-sample column. Usefeat_impact_'name'(signed) for the CPP-SHAP impact heatmap andmean_dif_'name'for the feature-value heatmap, not the group-levelfeat_importance(unsigned). The two are semantically opposite.Treating :meth:`~aaanalysis.CPPPlot.heatmap` as static: it is an instance method (
aa.CPPPlot().heatmap(...)).Over-generalizing one sample: SHAP values are sample- and model-specific. A high-impact feature for APP does not mean it generalizes to all substrates; cross-check against the group signature (P1).
Forgetting the ``pro`` extra:
ShapModelneeds shap (pip install aaanalysis[pro]).
Next step. P10: Validation (can I trust this?). Before reading a single sample’s explanation as biology, test the signature’s stability:
Repeated cross-validation: refit CPP +
ShapModelover random splits; do the SHAP rankings converge?Feature-drop tests: remove top-impact features one at a time; does performance degrade as expected?
Shuffled-label controls: refit on permuted labels; genuine impacts should collapse to noise.
Bootstrap confidence intervals: resample proteins within each class; do the attributions stay consistent?
See P10: Validation.