P6: Compositional vs positional
This protocol contrasts compositional and positional CPP
strategies. Once you have two labelled sets of sequences, a test
group (label=1) and a reference group (label=0), and you
want CPP to tell you what physicochemically distinguishes them, there
is one design choice that quietly governs everything: how finely
should each feature be localized? A CPP feature is always a
Part-Split-Scale triple, and it is the split that decides
whether a scale is averaged over a whole part (composition-like,
position-agnostic) or over specific sub-regions / positions
(location-resolved). You never set a strategy= switch: the strategy
emerges from the split_kws recipe you hand to CPP.
This protocol is a determinant-discovery workflow: we contrast two groups and read out the signature of the test group, but here the focus is the shape of that signature. We show how to write each recipe deliberately and how the choice maps onto the prediction level.
Composition answers “what” (how much of a property is present across
a part), position answers “where” (at which residues the property
differs). Compositional CPP = a single whole-part average,
Segment(1,1); positional CPP = sub-segments (n_split_max>1) plus
Pattern / PeriodicPattern. As a rule of thumb: compositional ~
protein level (SEQ_*), positional ~ residue level
(AA_*), and domain level (DOM_*) happily uses both.
When to use it. Use this protocol when you already have two labelled sets of sequences and need to decide how finely your CPP features should be localized before you commit to a signature. The unit of comparison stays fixed by your prediction level: it is only the locality of the split that you are tuning here.
Question |
Strategy |
Prediction level |
|---|---|---|
“Is one group globally enriched in a property?” (e.g. whole-chain composition) |
Compositional |
protein level
( |
“Where along the region does the property differ?” (e.g. a residue near a cleavage site) |
Positional |
residue level
( |
“Both: coarse composition **and* a location-specific signal in a defined sub-region”* |
Both |
domain level
( |
Compositional features are coarse and robust (few features, composition-like); positional features are fine and mechanistically actionable but more numerous. The strategy should match the biological resolution your question needs.
When not to use it. If you have not yet built df_parts or chosen
a prediction level, start with P1: CPP signature (and the parts/splits
engineering in P4: Engineer features) first; this protocol assumes
those are settled. It is also not a feature-selection step: here we
only shape the split grid; trimming the resulting signature down to a
compact, non-redundant set is the job of P7: Select & reduce features
(see Next step). And if your question genuinely is global (“is the
whole chain enriched?”), do not reach for a dense Pattern /
PeriodicPattern grid; that only inflates the feature count without
adding interpretive value.
Input. A df_seq with one row per protein and a binary label
column, the test group (label=1) versus the reference group
(label=0). It comes from an upstream dataset / sampling step (see
P1: CPP signature).
Here we work at the domain level with the bundled ``DOM_GSEC``
gamma-secretase dataset, so the unit of comparison is the
transmembrane-domain (TMD) part set, native ground for CPP. With
n=50 we load 2N = 100 rows (50 substrates + 50 non-substrates).
Each row carries entry, sequence, label, and the
domain-coordinate columns tmd_start, tmd_stop, jmd_n,
tmd, jmd_c that get_df_parts() consumes.
import aaanalysis as aa
aa.options["verbose"] = False
aa.options["random_state"] = 42
# Two labelled sets of sequences (label: 1 = substrate/test, 0 = reference)
# n=50 -> 2N=100 rows (50 substrates + 50 non-substrates)
df_seq = aa.load_dataset(name="DOM_GSEC", n=50)
labels = df_seq["label"].to_list()
aa.display_df(df=df_seq, n_rows=5)
| entry | gene | sequence | label | tmd_start | tmd_stop | jmd_n | tmd | jmd_c | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Q14802 | FXYD3 | MQKVTLGLLVFLAGF...PGETPPLITPGSAQS | 0 | 37 | 59 | NSPFYYDWHS | LQVGGLICAGVLCAMGIIIVMSA | KCKCKFGQKS |
| 2 | Q86UE4 | MTDH | MAARSWQDELAQQAE...SPKQIKKKKKARRET | 0 | 50 | 72 | LGLEPKRYPG | WVILVGTGALGLLLLFLLGYGWA | AACAGARKKR |
| 3 | Q969W9 | PMEPA1 | MHRLMGVNSTAAAAA...AIWSKEKDKQKGHPL | 0 | 41 | 63 | FQSMEITELE | FVQIIIIVVVMMVMVVVITCLLS | HYKLSARSFI |
| 4 | P53801 | PTTG1IP | MAPGVARGPTPYWRL...GLFKEENPYARFENN | 0 | 97 | 119 | RWGVCWVNFE | ALIITMSVVGGTLLLGIAICCCC | CCRRKRSRKP |
| 5 | Q8IUW5 | RELL1 | MAPRALPGSAVLAAA...EVPATPVKRERSGTE | 0 | 59 | 81 | NDTGNGHPEY | IAYALVPVFFIMGLFGVLICHLL | KKKGYRCTTE |
Run. The real entry point is
CPP(df_parts=..., split_kws=...).run(labels=...). CPP does
not take df_seq / labels directly: you first build sequence
parts with get_df_parts(), then pass a split_kws
recipe that encodes the strategy (see the CPP tutorial for the function
details). The only thing that differs between compositional and
positional is split_kws; the parts are identical.
# Build sequence parts once, shared by both strategies (TMD / JMD-N / JMD-C + composites)
sf = aa.SequenceFeature()
df_parts = sf.get_df_parts(df_seq=df_seq)
aa.display_df(df=df_parts, n_rows=5)
| tmd | jmd_n_tmd_n | tmd_c_jmd_c | |
|---|---|---|---|
| entry | |||
| Q14802 | LQVGGLICAGVLCAMGIIIVMSA | NSPFYYDWHSLQVGGLICAGVL | CAMGIIIVMSAKCKCKFGQKS |
| Q86UE4 | WVILVGTGALGLLLLFLLGYGWA | LGLEPKRYPGWVILVGTGALGL | LLLFLLGYGWAAACAGARKKR |
| Q969W9 | FVQIIIIVVVMMVMVVVITCLLS | FQSMEITELEFVQIIIIVVVMM | VMVVVITCLLSHYKLSARSFI |
| P53801 | ALIITMSVVGGTLLLGIAICCCC | RWGVCWVNFEALIITMSVVGGT | LLLGIAICCCCCCRRKRSRKP |
| Q8IUW5 | IAYALVPVFFIMGLFGVLICHLL | NDTGNGHPEYIAYALVPVFFIM | GLFGVLICHLLKKKGYRCTTE |
# --- Recipe A: COMPOSITIONAL (position-agnostic, whole-part average) ---
# A single Segment(1,1) per part -> the feature is a composition-like mean over the ENTIRE part.
split_kws_comp = sf.get_split_kws(split_types="Segment",
n_split_min=1,
n_split_max=1)
split_kws_comp
{'Segment': {'n_split_min': 1, 'n_split_max': 1}}
cpp = aa.CPP(df_parts=df_parts, split_kws=split_kws_comp)
df_feat_comp = cpp.run(labels=labels, n_filter=20) # compositional yields fewer candidates than positional
aa.display_df(df=df_feat_comp[["feature", "category", "subcategory", "abs_auc", "mean_dif", "positions"]], n_rows=10)
| feature | category | subcategory | abs_auc | mean_dif | positions | |
|---|---|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...1,1)-WOLS870103 | Others | PC 4 | 0.393000 | -0.079000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 2 | TMD_C_JMD_C-Seg...1,1)-ZIMJ680104 | Energy | Isoelectric point | 0.339000 | 0.056000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 3 | TMD_C_JMD_C-Seg...1,1)-AURR980112 | Conformation | α-helix | 0.335000 | 0.069000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 4 | TMD_C_JMD_C-Seg...1,1)-WILM950103 | Polarity | Hydrophobicity (interface) | 0.288000 | -0.059000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 5 | TMD_C_JMD_C-Seg...1,1)-GEOR030108 | Structure-Activity | Stability (helix-coil) | 0.287000 | 0.076000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 6 | TMD_C_JMD_C-Seg...1,1)-CHOP780212 | Conformation | β-sheet (C-term) | 0.282000 | -0.067000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 7 | TMD_C_JMD_C-Seg...1,1)-JACR890101 | Polarity | Hydrophobicity (surrounding) | 0.281000 | -0.057000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 8 | TMD_C_JMD_C-Seg...1,1)-JANJ780101 | ASA/Volume | Accessible surface area (ASA) | 0.278000 | 0.058000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 9 | TMD_C_JMD_C-Seg...1,1)-FUKS010109 | Composition | AA composition | 0.277000 | 0.061000 | 21,22,23,24,25,...,36,37,38,39,40 |
| 10 | JMD_N_TMD_N-Seg...1,1)-KARP850101 | Structure-Activity | Flexibility | 0.270000 | 0.057000 | 1,2,3,4,5,6,7,8...,16,17,18,19,20 |
# --- Recipe B: POSITIONAL (location-resolved sub-regions / positions) ---
# Sub-segments (n_split_max>1) PLUS Pattern and PeriodicPattern -> features resolve to WHERE in the part.
split_kws_pos = sf.get_split_kws(split_types=["Segment", "Pattern", "PeriodicPattern"],
n_split_min=1,
n_split_max=5,
steps_pattern=[3, 4],
steps_periodicpattern=[3, 4])
split_kws_pos
{'Segment': {'n_split_min': 1, 'n_split_max': 5},
'Pattern': {'steps': [3, 4], 'n_min': 2, 'n_max': 4, 'len_max': 15},
'PeriodicPattern': {'steps': [3, 4]}}
cpp = aa.CPP(df_parts=df_parts, split_kws=split_kws_pos)
df_feat_pos = cpp.run(labels=labels, n_filter=30)
aa.display_df(df=df_feat_pos[["feature", "category", "subcategory", "abs_auc", "mean_dif", "positions"]], n_rows=10)
| feature | category | subcategory | abs_auc | mean_dif | positions | |
|---|---|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...2,3)-QIAN880106 | Conformation | α-helix | 0.387000 | 0.121000 | 27,28,29,30,31,32,33 |
| 2 | TMD_C_JMD_C-Pat...,12)-ROBB760109 | Conformation | β-turn (N-term) | 0.377000 | -0.127000 | 21,25,28,32 |
| 3 | TMD_C_JMD_C-Seg...4,5)-ZIMJ680104 | Energy | Isoelectric point | 0.373000 | 0.220000 | 33,34,35,36 |
| 4 | TMD_C_JMD_C-Seg...4,5)-WOLS870103 | Others | PC 4 | 0.370000 | -0.218000 | 33,34,35,36 |
| 5 | TMD_C_JMD_C-Seg...3,4)-FAUJ880104 | Shape | Side chain length | 0.366000 | 0.205000 | 31,32,33,34,35 |
| 6 | TMD_C_JMD_C-Seg...2,3)-WOLS870103 | Others | PC 4 | 0.365000 | -0.154000 | 27,28,29,30,31,32,33 |
| 7 | TMD_C_JMD_C-Seg...4,5)-ONEK900101 | Others | Unclassified (Others) | 0.364000 | 0.097000 | 33,34,35,36 |
| 8 | TMD_C_JMD_C-Seg...4,5)-FINA910103 | Conformation | α-helix (C-cap) | 0.362000 | 0.264000 | 33,34,35,36 |
| 9 | TMD_C_JMD_C-Pat...,15)-QIAN880107 | Conformation | α-helix | 0.359000 | 0.158000 | 25,28,32,35 |
| 10 | TMD_C_JMD_C-Pat...,15)-MUNV940101 | Energy | Free energy (folding) | 0.358000 | -0.097000 | 24,28,32,35 |
# Rank each signature by tree-based feature importance (needed by feature_map).
# One clearly-named TreeModel per strategy keeps the comp/pos blocks parallel.
tm = aa.TreeModel()
X_comp = sf.feature_matrix(features=df_feat_comp["feature"], df_parts=df_parts)
df_feat_comp = tm.fit(X_comp, labels=labels).add_feat_importance(df_feat=df_feat_comp)
tm = aa.TreeModel()
X_pos = sf.feature_matrix(features=df_feat_pos["feature"], df_parts=df_parts)
df_feat_pos = tm.fit(X_pos, labels=labels).add_feat_importance(df_feat=df_feat_pos)
aa.display_df(df=df_feat_pos[["feature", "subcategory", "mean_dif", "abs_auc", "feat_importance"]], n_rows=6)
| feature | subcategory | mean_dif | abs_auc | feat_importance | |
|---|---|---|---|---|---|
| 1 | TMD_C_JMD_C-Seg...2,3)-QIAN880106 | α-helix | 0.121000 | 0.387000 | 8.225000 |
| 2 | TMD_C_JMD_C-Pat...,12)-ROBB760109 | β-turn (N-term) | -0.127000 | 0.377000 | 5.570000 |
| 3 | TMD_C_JMD_C-Seg...4,5)-ZIMJ680104 | Isoelectric point | 0.220000 | 0.373000 | 3.317000 |
| 4 | TMD_C_JMD_C-Seg...4,5)-WOLS870103 | PC 4 | -0.218000 | 0.370000 | 3.021000 |
| 5 | TMD_C_JMD_C-Seg...3,4)-FAUJ880104 | Side chain length | 0.205000 | 0.366000 | 4.483000 |
| 6 | TMD_C_JMD_C-Seg...2,3)-WOLS870103 | PC 4 | -0.154000 | 0.365000 | 3.341000 |
import matplotlib.pyplot as plt
import seaborn as sns
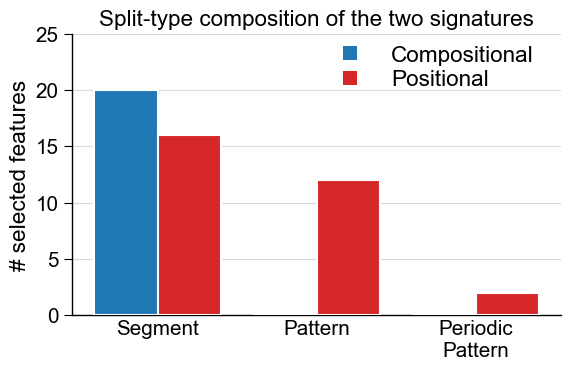
# Feature id is PART-SPLIT-SCALE; the SPLIT field's head names the split type.
def split_type_counts(df_feat):
types = df_feat["feature"].str.split("-").str[1].str.split("(").str[0]
return types.value_counts()
order = ["Segment", "Pattern", "PeriodicPattern"]
counts_comp = split_type_counts(df_feat_comp).reindex(order, fill_value=0)
counts_pos = split_type_counts(df_feat_pos).reindex(order, fill_value=0)
aa.plot_settings(font_scale=0.9, weight_bold=False)
colors = aa.plot_get_clist(n_colors=2)
fig, ax = plt.subplots(figsize=(6, 4))
x = range(len(order))
w = 0.4
ax.bar([i - w / 2 for i in x], counts_comp.values, width=w, label="Compositional",
color=colors[0], edgecolor="white", zorder=3)
ax.bar([i + w / 2 for i in x], counts_pos.values, width=w, label="Positional",
color=colors[1], edgecolor="white", zorder=3)
ax.set_xticks(list(x))
ax.set_xticklabels(["Segment", "Pattern", "Periodic\nPattern"])
ax.set_ylabel("# selected features")
ax.set_title("Split-type composition of the two signatures")
ax.set_ylim(0, max(counts_comp.max(), counts_pos.max()) * 1.25)
ax.grid(axis="y", color="0.85", zorder=0)
ax.tick_params(axis="x", length=0)
sns.despine()
aa.plot_legend(dict_color={"Compositional": colors[0], "Positional": colors[1]},
n_cols=1, x=0.5, y=1.0, marker="s", marker_size=10)
plt.tight_layout()
plt.show()
Output. Both strategies return the same df_feat signature
schema (one row per selected feature). Key columns:
feature: thePart-Split-Scaleid (where x how x which property). The Split field is what differs: compositional ids carry onlySegment(1,1); positional ids carrySegment(i,n),Pattern(...), orPeriodicPattern(...).category/subcategory: the AAontology property group.abs_auc: effect size / group-separation strength.mean_dif: mean difference (test minus reference); the sign gives the direction.positions: the residue positions the feature averages over (a single broad span for compositional; tight, specific positions for positional).feat_importance: tree-based importance used to rank the signature.
The bar plot above makes the strategic difference concrete: the
compositional signature is all ``Segment`` (whole-part means), while
the positional signature mixes Segment sub-regions, Pattern, and
PeriodicPattern. Read the two feature maps below as rows =
properties, columns = positions along the parts, colour = direction x
strength.
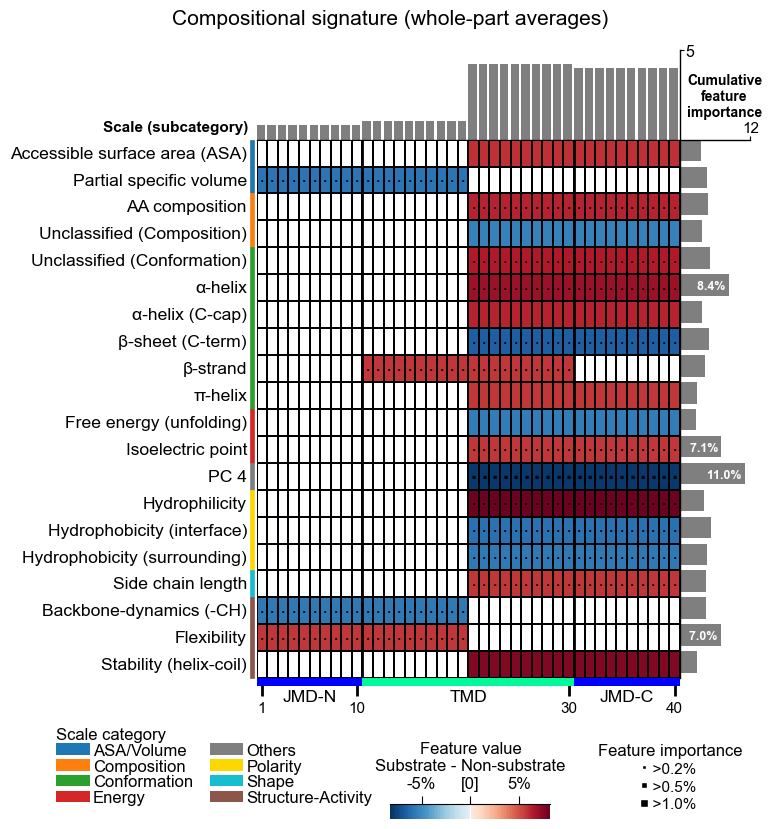
aa.plot_settings(font_scale=0.7, weight_bold=False)
cpp_plot = aa.CPPPlot()
cpp_plot.feature_map(df_feat=df_feat_comp, name_test="Substrate", name_ref="Non-substrate")
plt.gcf().suptitle("Compositional signature (whole-part averages)", y=1.02)
plt.show()
aa.plot_settings(font_scale=0.7, weight_bold=False)
cpp_plot = aa.CPPPlot()
cpp_plot.feature_map(df_feat=df_feat_pos, name_test="Substrate", name_ref="Non-substrate")
plt.gcf().suptitle("Positional signature (location-resolved)", y=1.02)
plt.show()
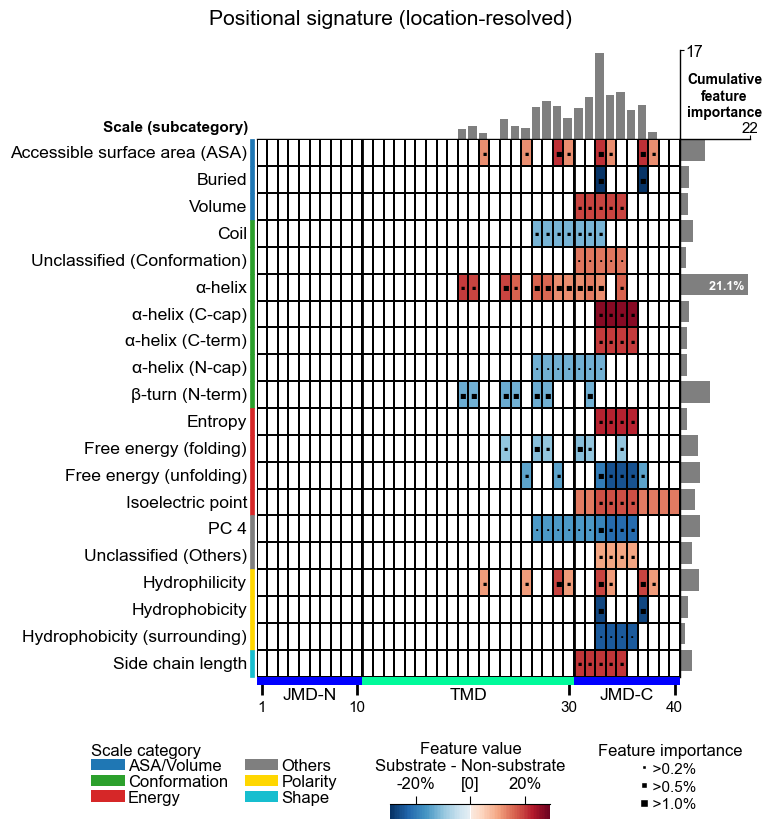
How to interpret. Notice how the biological reading mirrors the
strategy you chose. The Part- prefix below is schematic: it stands
for whichever part actually wins the filter (in this DOM_GSEC run
that happens to be the composite TMD_C_JMD_C, as the displayed
df_feat shows), so read the ids for their Split field, not the
literal part name:
Strategy |
Feature id looks like |
Biological reading ( gamma-secretase example) |
Best prediction level |
|---|---|---|---|
** Compositional** |
|
“Substrates are richer in small hydrophobic residues across the whole part.”: coarse, co mposition-like, robust. |
protein level |
Positional |
|
“Substrates differ in a charged-residue propensity at specific positions near the cleavage site (TMD centre).”: fine, loc ation-specific, mechanistically actionable. |
residue level |
Both |
mix of the above |
domain-level tasks benefit from coarse composition and local position signal in one signature. |
domain level |
In short: compositional answers “how much” (global property level); positional answers “where” (location of the discriminating property). For mechanism (pinning a signal to a cleavage-adjacent residue), the positional strategy is the more actionable one.
Key takeaways
The strategy is not a parameter; it emerges from ``split_kws``.
Segment(1,1)alone is compositional; addingn_split_max>1/Pattern/PeriodicPatternmakes it positional.Both recipes return the same ``df_feat`` signature schema: only the
Splitfield of thefeatureid (and how tight thepositionsspan is) tells the two apart.Match locality to the question: compositional ~ protein level (the “what”), positional ~ residue level (the “where”), domain level uses both.
Common mistakes.
Using a positional recipe for a protein-level task. Whole-chain composition questions only need
Segment(1,1); a largePattern/PeriodicPatterngrid over a long chain explodes the feature count for no interpretive gain.Using a compositional recipe for a residue-level task. A single whole-part average cannot localize a site-specific signal, you lose exactly the where the question is about.
Not matching strategy to prediction level. Remember the mapping: compositional ~ protein, positional ~ residue, domain uses both. There is no
strategy=parameter; the strategy emerges fromsplit_kws.Forgetting ``feat_importance``.
feature_map()is an instance method and needs afeat_importancecolumn; add it withadd_feat_importance()afterfit(skipping this raises infeature_map).
Next step. Both signatures here were capped with a plain
n_filter, so they may still carry redundant features. To turn a raw
signature into a compact, non-redundant feature set (redundancy
reduction plus tree-based feature selection), continue with P7: Select
& reduce features.